


Renal cell tumors comprise both benign – oncocytoma – and malignant – clear cell renal cell carcinoma, papillary renal cell carcinoma, and chromophobe renal cell carcinoma – entities. Since the differential diagnosis among renal cell tumors is sometimes difficult on clinical, imaging and pathological grounds, and prognosis is quite dissimilar, epigenetic-based diagnostic biomarkers, specially promoter methylation, might be useful for accurate diagnosis and therapeutic planning.
Materials and methodsEpiTect Methyl II PCR Array was used to screen methylation status of 22 genes, involved in epithelial to mesenchymal transition. Quantitative real-time methylation specific polymerase chain reaction was performed for candidate gene validation, and methylation levels of renal cell tumors subtypes and normal kidney were determined and compared.
ResultsMST1R promoter methylation level was significantly higher in clear cell renal cell carcinoma (median: 5367) compared to other renal cell tumors (median: papillary renal cell carcinoma – 1084, chromophobe renal cell carcinoma – 1023, oncocytoma – 1337) and normal kidney (median: 1125), allowing for accurate discrimination from other renal cell tumors with high sensitivity (>96.7%) and specificity (86.7%).
ConclusionQuantitative MST1R promoter methylation may be useful as biomarker for accurate diagnosis of clear cell renal cell carcinoma in problematic cases.
Os tumores de células renais englobam neoplasias benignas – oncocitoma – e malignas – carcinoma de células renais de tipo célula clara, papilar e de células cromófobas. Uma vez que o diagnóstico diferencial entre eles não é linear do ponto de vista clínico, imagiológico e patológico, e que o prognóstico de cada subtipo é distinto, o desenvolvimento de biomarcadores de diagnóstico com base em alterações epigenéticas, nomeadamente a metilação do promotor de genes, poderá ser útil para o diagnóstico e planeamento do tratamento.
Material e métodosA presença de metilação do promotor de 22 genes envolvidos na transição epitélio-mesênquima foi avaliada através da plataforma comercial EpiTectMethylIIPCR Array. Os resultados foram validados, no gene candidato, por “polymerase chain reaction” quantitativo em tempo real específico para metilação. Os níveis de metilação de cada tipo histológico de tumores de células renais e de tecido renal normal foram calculados e comparados.
ResultadosO nível de metilação do promotor do gene MST1R foi significativamente mais elevado nos carcinomas de células renais de tipo célula clara (mediana: 5.367) comparativamente com carcinomas de células renais papilares (mediana: 1.084), carcinomas de células renais de células cromófobas (mediana: 1.023), oncocitomas (mediana: 1.337) e rim normal (mediana: 1.125), permitindo identificar carcinomas de células renais de tipo célula clara com elevada sensibilidade (> 96,7%) e especificidade (86,7%).
ConclusãoO nível de metilação do promotor do gene MST1R poderá constituir um biomarcador útil para o diagnóstico de carcinomas de células renais de tipo célula clara em casos problemáticos.
Renal cancer has an estimated age-standardized incidence and mortality rate respectively of 4.4/100,000 and 1.8/100,000 worldwide.1 In Europe, renal cancer incidence ranks seventh and eighth among all non-cutaneous malignant neoplasms, and in North America kidney and renal pelvis cancers account for 65,150 estimated new cases and 13,680 deaths in both genders in 2013, with men being more affected than women.2 Besides the classical presentation as metastatic disease, mostly because kidney cancer is asymptomatic at its earliest stages, an increasingly higher number of tumors are being incidentally discovered as small masses, frequently posing diagnostic challenges in what concerns the distinction between benign and malignant tumors.3
Renal cell tumors are the most common neoplasms of the kidney (80–85%), followed by urothelial carcinoma of the renal pelvis (15–20%).4 Renal cell tumors (RCTs) are heterogeneous at the genetic, morphological and clinical levels, comprising both benign and malignant entities.5,6 Underscoring the high heterogeneity of RCT, malignant tumors – renal cell carcinomas (RCC) – might have different responses to novel targeted therapies.7 The most frequent malignant RCT is clear cell renal cell carcinoma (ccRCC), followed by papillary renal cell carcinoma (pRCC) and chromophobe renal cell carcinoma (chRCC), accounting respectively for 70%, 10–15% and 5% of all RCT.4 Oncocytoma, the most common benign tumor, accounts for approximately 5% of all RCT.5
Histological subtyping provides relevant prognostic information, independent from tumor pathological stage and grade: ccRCC is the most aggressive subtype, displaying lower cancer-specific survival than pRCC and chRCC, whereas no differences in cancer specific survival are apparent between pRCC and chRCC.8,9
Usually, information on histological subtype is available only after pathological evaluation of the nephrectomy specimen, and thus this valuable diagnostic and prognostic data cannot be taken into account for planning the best time for surgery, especially in cases of small renal tumors (<4cm) that should benefit from nephron sparing therapy.3,10 For these small tumors, limitations of imagiological techniques and core biopsy histopathological examination preclude accurate characterization of some lesions, seriously limiting the pre-operative information about the biological behavior of the tumor available for the urologist.3,10 Moreover, in the setting of active surveillance protocols,11,12 the development of novel diagnostic biomarkers, preferentially minimally- or non-invasive, is of paramount importance.
The association between both genetic and epigenetic alterations and renal cancer development is widely acknowleged.4,13,14 Additionally, specific genetic and/or epigenetic alterations were found to be characteristic of certain tumor types, and therefore may act as specific diagnostic biomarkers.13,14 Epigenetic biomarkers, such as methylation status of a gene promoter (promoter hipermethylation), which is associated with repression of gene expression, may be detected in the patient's serum or urine.15,16 Thus, it may constitute important pre-operative non-invasive diagnostic tools and/or useful ancillary test for the diagnosis of renal masses through fine needle aspiration cytology or tissue biopsy, allowing for the discrimination between benign and malignant RCT and/or for the identification of more aggressive malignant tumors.
Tumor aggressiveness in the form of local invasion and acquisition of metastatic potential has been associated with epithelial to mesenchymal transition (EMT).17 Key EMT effectors were described to be regulated by epigenetic mechanisms,18,19 and EMT-related genes were found to be deregulated in renal cell tumors.14 Thus, epigenetic deregulation of cellular pathways involved in EMT might account for differences in RCT's aggressiveness, and provide useful diagnostic and prognostic biomarkers.
Therefore, we aimed to systematically evaluate the methylation status of EMT-related genes in the four most frequent RCT subtypes and investigate their potential use as clinically relevant diagnostic and prognostic tools.
MethodsPatients, sample collection and DNA extractionTumor tissue from 120 total or partial nephrectomy specimens performed between 2003 and 2007 (30 of each ccRCC, pRCC, chRCC and oncocytoma) (Fig. 1), and morphologically normal kidney (cortical) tissue from 9 nephrectomy specimens for upper urinary tract neoplasia and 12 renal biopsies performed in the diagnostic work-up of renal dysfunction (without neoplasia or inflammation), were included in this study. All procedures were performed at the Portuguese Oncology Institute – Porto (Portugal), after informed consent was obtained.
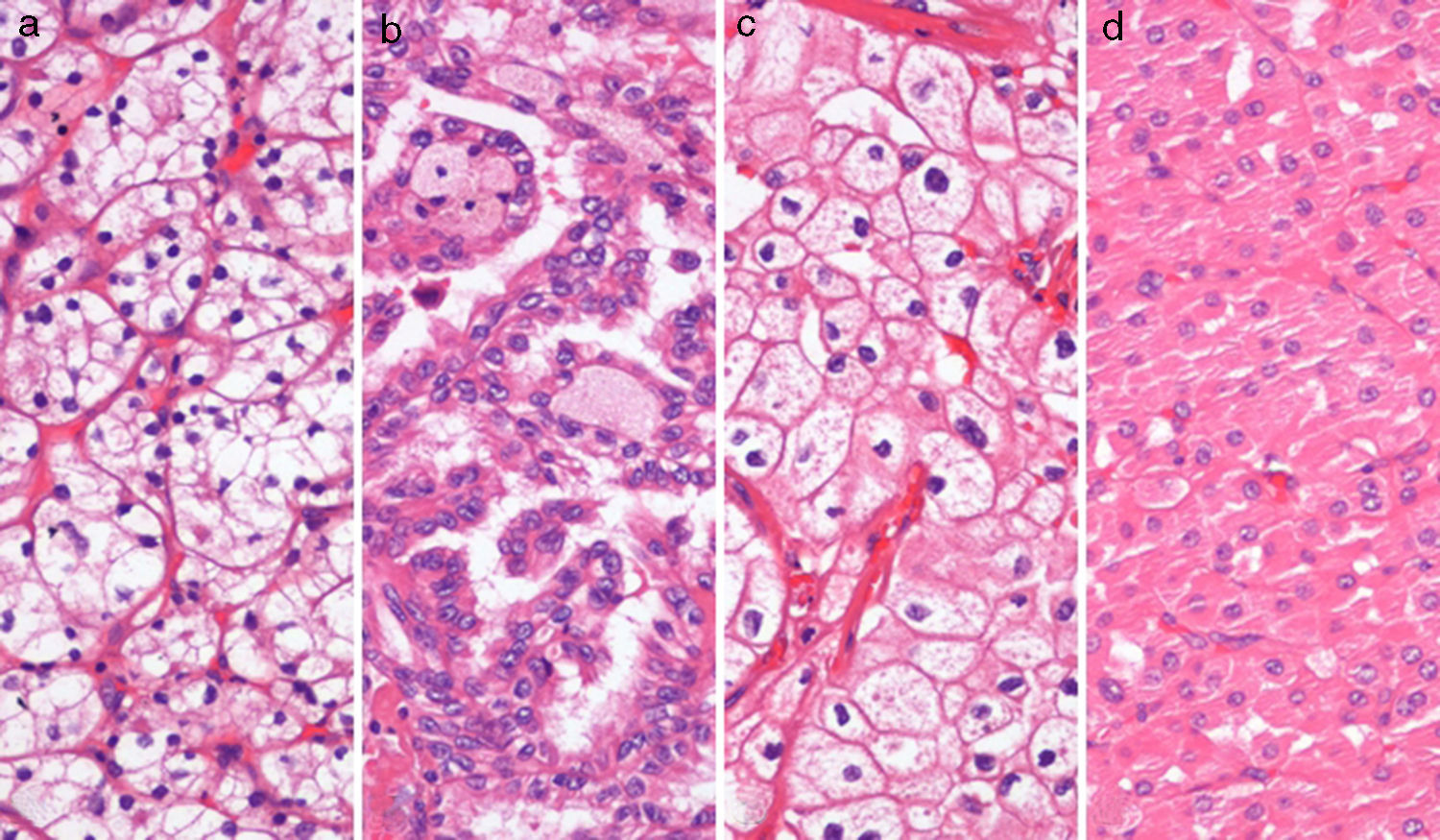
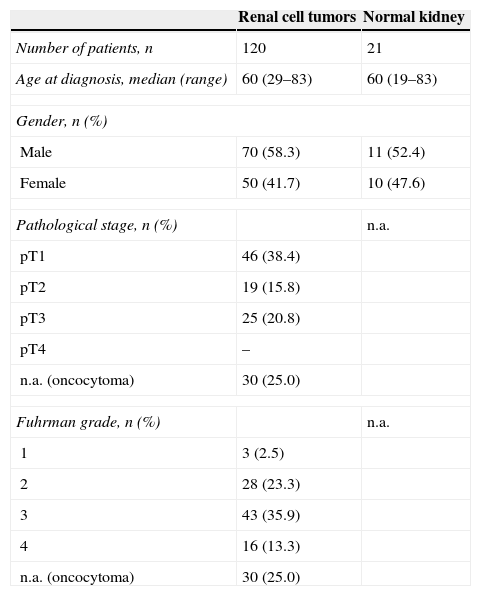
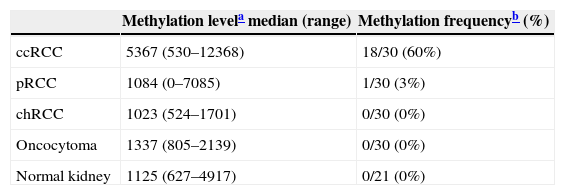
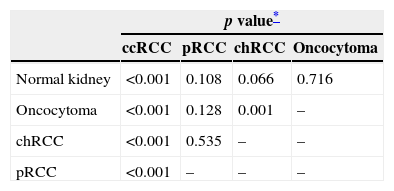
 morphology: (a) clear cell renal cell carcinoma (ccRCC), (b) papillary renal cell carcinoma (pRCC), (c) chromophome renal cell carcinoma (chRCC) and (d) oncocytoma (hematoxylin and eosin staining 200×).")
Tumor tissue samples were obtained immediately after surgery and snap-frozen. Renal biopsies were frozen as part of the routine protocol. All samples were stored at −80°C and subsequently cut in a cryostat. The presence and extent of tumor were evaluated by H&E stains, to ensure at least 70% of tumor in the samples.
Genomic DNA was extracted as previously described.20 Briefly, DNA was digested overnight with proteinase K (20mg/mL) in the presence of 10% SDS at 55°C, extracted with phenolchloroform and precipitated with 100% ethanol.
Tumor classification (WHO), grading (Fuhrman) and staging (TNM) were routinely assessed for all tumor cases in formalin-fixed paraffin-embedded tissue.5,21
Relevant clinical data were collected from clinical charts. This study was approved by the Institutional Review Board (Comissão de Ética para a Saúde) of Portuguese Oncology Institute – Porto, Portugal (CES518/2010).
EpiTect Methyl II qPCR arrayThe EpiTect Methyl II PCR Array (SABiosciences, Qiagen, Frederick, MD, USA) was used as a screening method to evaluate the promoter methylation status of EMT-related genes in 20 samples (4 ccRCC; 4 pRCC; 6 chRCC; 6 oncocytoma). The EMT commercial assay (cat. no. 524EAHS-901ZA-24) includes 22 genes: CDH1, CTNNAL1, DSC2, DSP, EPCAM, GAB1, KRT19, KRT7, MAP3K5, MST1R, NID2, OCLN, PLEK2, PLSCR1, PPPDE2, PTP4A1, RGS2, SEH1L, SMAD4, TGIF1, TSPAN13 and YES1.
Assays were performed according to the manufacturer's instructions. Briefly, equal amounts of genomic DNA were incubated overnight at 37°C with a DNA methylation-sensitive restriction enzyme, which digests unmethylated DNA; with a DNA methylation-dependent restriction enzyme that digests methylated DNA; with both enzymes; and without enzyme added/“mock” (Methyl-Profiler DNA Methylation Enzyme Kit, SABiosciences). The enzyme was inactivated at 65°C for 20min and, after digestion, the remaining DNA was quantified by real-time PCR using pre-designed primers to the promoter region. DNA amplification was carried on a 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA), at 95°C for 10min followed by 40 cycles of 97°C for 15s and 72°C for 1min. PCR product was marked with SYBR® Green and Ct values were obtained. The analysis was performed using a SABiosciences Excel-Based Data Analysis Template, and the percent of hypermethylated DNA was obtained by comparing the amount of DNA in each digest with that of a mock digest, representing the fraction of input DNA containing at least two methylated CpG sites in the targeted gene region.
Bisulfite treatmentSodium bisulfite converts unmethylated cytosine residues to uracil, whereas methylated cytosine residues remained as such. It was performed with EZ DNA Methylation-Gold Kit (Zymo Research), according to the manufacturer's instructions, in a total of 120 tumor samples and 21 morphologically normal kidney tissues, as mentioned above.
Quantitative MSPQuantitative real-time polymerase chain reaction (qMSP) was performed for all the tested samples after DNA bisulfite treatment.
Primers were designed to amplify methylated bisulfite converted complementary sequences of MST1R promoter using Methyl Primer Express v 1.0 (Applied Biosystems, Foster City, CA, USA). Primer sequences used for Macrophage-stimulating-1 receptor (MST1R) [GenBank: NM_002447] were 5′GCGAGGATTGGTAGTGTTC3′ (forward) and 5′TTCTATCGCCCTCGTAAATC3′ (reverse). A reference gene (β-actin) was used to normalize for DNA input in each sample.
QMSP analysis was performed using an 7500 Real-time PCR system (Applied Biosystems, Foster City, CA, USA), in a reaction volume of 20μL consisting of 10μL of SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA), 7μL of H2O, 0.5μL of forward primer, 0.5μL of reverse primer and 2μL of bisulfate-modified DNA. Each sample was run in triplicate and “no template controls” were included in each plate as a control for contamination. A calibration curve to quantify the amount of fully methylated alleles in each reaction was constructed with serial dilutions (1:5) of bisulfite converted universally methylated DNA at all CpGs (CpGenome Universal Methylated DNA; Millipore, Billerica, MA). The amplification reaction was carried out at 95°C for 2min followed by 50 cycles of 95°C for 15s and annealing temperature (60°C) for 1min, followed by melting curve analysis.
Relative levels of methylated promoter DNA in each sample were determined by dividing the mean quantity obtained by qMSP analysis for each gene for the respective value of the internal reference gene (ACTB). This value was multiplied by 1000 for easy tabulation (methylation level=target gene/reference gene×1000).
Statistical analysisThe frequency of methylated samples was determined for the all four RCT types, considering the highest value determined in the normal kidney tissue as the cutoff. Both median and interquartile ranges of methylation levels were also determined. Differences in methylation levels among RCT subtypes were analyzed using Kruskal–Wallis non-parametric ANOVA followed by Mann–Whitney U test, when appropriate. p values in multiple comparisons were adjusted according to the Bonferroni method. Statistical significance level was set at p<0.05 (two-sided). Disease specific survival and disease free survival curves (Kaplan–Meier with log-rank test) were computed for histological subtype and MST1R promoter methylation levels. Statistical analysis was performed using IBM® SPSS® Statistics for Windows, version 22.0 (SPSS, Chicago, IL, USA).
ResultsPromoter methylation screening with EpiTect Methyl II PCR ArrayFive out of the 22 EMT-related genes tested – CDH1, DSC2, KRT7, MST1R and TSPAN13 – displayed hypermethylation at the respective promoter regions. Among these, MST1R depicted the highest percent of hypermethylated DNA – >90% – meaning that at least two CpG sites in the targeted gene region were methylated in more than 90% of input DNA sample. Thus, MST1R promoter metylation was chosen for subsequent validation.
MST1R promoter methylation analysis by qMSPRelevant clinical and pathological data of the 120 tumors included in this study are depicted in Table 1. No statistically significant associations between MST1R promoter methylation levels and pathological stage were found (p=0.06). However, MST1R methylation levels of Fuhrman grade 3 tumors (median: 1451; range: 401–12,367) were higher than those with Fuhrman grade 4 (median: 938, range: 718–7085), and this difference attained statistical significance (p=0.011).
Clinical and pathological features of patients included in this study.
| Renal cell tumors | Normal kidney | |
|---|---|---|
| Number of patients, n | 120 | 21 |
| Age at diagnosis, median (range) | 60 (29–83) | 60 (19–83) |
| Gender, n (%) | ||
| Male | 70 (58.3) | 11 (52.4) |
| Female | 50 (41.7) | 10 (47.6) |
| Pathological stage, n (%) | n.a. | |
| pT1 | 46 (38.4) | |
| pT2 | 19 (15.8) | |
| pT3 | 25 (20.8) | |
| pT4 | – | |
| n.a. (oncocytoma) | 30 (25.0) | |
| Fuhrman grade, n (%) | n.a. | |
| 1 | 3 (2.5) | |
| 2 | 28 (23.3) | |
| 3 | 43 (35.9) | |
| 4 | 16 (13.3) | |
| n.a. (oncocytoma) | 30 (25.0) | |
n.a., not applicable.
To categorize samples as methylated or unmethylated, a cutoff value was chosen based on the highest methylation ratio value obtained for the respective normal/control samples, ensuring perfect specificity of the assay. We found that in ccRCC the methylation frequency was 60% and, in the remaining histological subtypes, methylation frequency was very low (1%) in pRCC, and absent in chRCC and oncocytoma. The most aggressive RCC subtype, ccRCC, displayed higher MST1R methylation levels compared to pRCC, chRCC and oncocytomas (Table 2), and this was statistically significant (Table 3). MST1R methylation levels of chRCC also significantly differed from those of oncocytoma (p=0.008, Table 3).
MST1R methylation levels and frequencies among normal kidney tissues and different renal cell tumor subtypes.
| Methylation levela median (range) | Methylation frequencyb (%) | |
|---|---|---|
| ccRCC | 5367 (530–12368) | 18/30 (60%) |
| pRCC | 1084 (0–7085) | 1/30 (3%) |
| chRCC | 1023 (524–1701) | 0/30 (0%) |
| Oncocytoma | 1337 (805–2139) | 0/30 (0%) |
| Normal kidney | 1125 (627–4917) | 0/21 (0%) |
ccRCC, clear cell renal cell carcinoma; pRCC, papillary renal cell carcinoma; chRCC, chromophobe renal cell carcinoma.
Comparison of the distribution of MST1R methylation levels among renal cell tumor subtypes and normal kidney.
| p value* | ||||
|---|---|---|---|---|
| ccRCC | pRCC | chRCC | Oncocytoma | |
| Normal kidney | <0.001 | 0.108 | 0.066 | 0.716 |
| Oncocytoma | <0.001 | 0.128 | 0.001 | – |
| chRCC | <0.001 | 0.535 | – | – |
| pRCC | <0.001 | – | – | – |
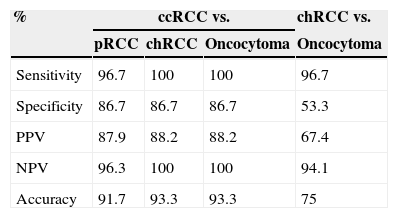
MST1R methylation levels allowed for the discrimination between ccRCC and the remainder RCT – pRCC, chRCC and oncocytoma – with high sensitivity (96.7%, 100%, 100%, respectively) and specificity (86.7% for the three comparisons), as well as between chRCC and oncocytoma, with 96.7% sensitivity and 53.3% specificity (Table 4).
Validity estimates for MST1R promoter methylation level in different settings.
| % | ccRCC vs. | chRCC vs. | ||
|---|---|---|---|---|
| pRCC | chRCC | Oncocytoma | Oncocytoma | |
| Sensitivity | 96.7 | 100 | 100 | 96.7 |
| Specificity | 86.7 | 86.7 | 86.7 | 53.3 |
| PPV | 87.9 | 88.2 | 88.2 | 67.4 |
| NPV | 96.3 | 100 | 100 | 94.1 |
| Accuracy | 91.7 | 93.3 | 93.3 | 75 |
PPV, positive predictive value; NPV, negative predictive value; ccRCC, clear cell renal cell carcinoma; pRCC, papillary renal cell carcinoma; chRCC, chromophobe renal cell carcinoma.
MST1R promoter methylation levels did not associate with cancer-specific survival (p=0.21) nor disease-free survival (p=0.091).
DiscussionMacrophage-stimulating-1 receptor (MST1R), also known as Ron (Recepteur d’Origine Nantais) is a receptor tyrosine kinase, member of the MET proto-oncogene family. MST1R binds macrophage stimulating protein (MSP) and is found in macrophages, epithelial cells, osteoclasts and hematopoietic cells.22–24 In normal epithelial cells, MST1R is involved not only in cell proliferation and survival (inhibition of apoptosis) but also in integrin-dependent cell adhesion and motility.22–24 MSP/MST1R signaling deregulation was found in cancer cells,25–27 and MST1R altered expression was reported in several human neoplasms, including hepatocellular,28 breast,29 colon,30 lung,31 ovarian,32 nasopharyngeal33 and bladder34 cancer, but not in renal cancer. Several oncogenic MST1R variants have been described,25–27 and hypermethylation of a specific region of MST1R promoter was reported to be associated with the transcription of an oncogenic MST1R variant,35 but MST1R promoter methylation had not been previously described in RCTs.
In this study, we showed that quantitative MST1R promoter methylation is a common feature of ccRCC, and its levels are significantly higher than those observed in pRCC, chRCC, oncocytoma and normal renal tissue, suggesting not only a role in renal tumorigenesis but also a specific implication in the genesis of the clear cell type. Furthermore, the distribution of MST1R promoter methylation levels allowed for accurate discrimination of ccRCC from other RCT, a finding that is of particular diagnostic relevance. It had been previously reported that the promoter methylation level of a panel of three genes – CDH1, PTGS2 and RASSF1A – could discriminate the four main histological types of RCT, although with limited sensitivity.36 Thus, either alone or in conjunction with that gene panel, MST1R promoter methylation might provide a useful ancillary tool for the histopathological or cytopathological evaluation of suspicious renal masses.
Although previous studies on RCT identified promoter hypermethylation of several genes37,38 using genome-wide promoter methylation analysis, data validation in an independent clinical series of samples is mostly lacking, impairing the evaluation of its usefulness as diagnostic biomarkers. On the other hand, most studies are focused in ccRCC, the most common RCT subtype, but increasing attention must be paid to diagnostic biomarkers which allow for the discrimination among RCTs, especially chRCC and oncocytoma, as diagnosis carries quite different prognosis and, eventually, therapeutic options. Global methylation profiles of chRCC and oncocytoma have been published,39 but again without validation in independent set of clinical samples. Although MST1R promoter methylation surfaced in the array as promising biomarker, we further extended our analysis to primary RCT and normal kidney tissues, to assess its potential clinical usefulness. Furthermore, this analysis allowed for the estimation of the sensitivity and specificity of this novel epigenetic-based biomarker, which depicted high diagnostic performance. However, because it does not allow for the coverage of all relevant differential diagnostic problems in RCT, the integration with other biomarkers, such as the microRNA that we recently demonstrated to distinguish among RCT subtypes,40 might be relevant, especially in the setting of patient selection for surveillance protocols. Importantly, these molecular techniques might be performed in clinical samples obtained by non- or minimally-invasive procedures, such as urine or serum,16 or serve as ancillary tools for fine needle aspiration cytology (FNAC), as previously shown.40
This study was performed in tumor tissue after nephrectomy, which limits the scope of this new diagnostic biomarker. Further validation of MST1R promoter methylation in urine and/or serum samples is warranted. Additionally, at present, the precise role of MST1R promoter methylation in ccRCC carcinogenesis is not known, nor is its relevance for the development of non-ccRCC. Elucidation of the biological role of MST1R in renal carcinogenesis might allow for the development of novel therapeutic strategies, as tyrosine kinase inhibitors that act on MST1R might modulate its effects on neoplastic cells.41,42
ConclusionsMST1R methylation levels might be a useful diagnostic biomarker, allowing for the discrimination of ccRCC from other RCT and normal renal tissue, with high sensitivity and specificity. Further studies are warranted to independently assess MST1R promoter methylation level as a new diagnostic biomarker for RCTs, and also to illuminate the role of MST1R promoter methylation in renal carcinogenesis.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
FundingBolsa de Investigação da Associação Portuguesa de Urologia (2010) & CIPOP 4 – 2008. ASP-L is supported by FCT - Fundação para a Ciência e Tecnologia-SFRH/SINTD/94217/2013.
