


Lymphangioleiomyomatosis (LAM) is a rare progressive disease that has gone largely unrecognised by paediatricians, even by paediatric pulmonologists. We report a paediatric case of sporadic LAM (S-LAM) that first manifested as recurrent spontaneous pneumothorax.
We report the case of a 17-year-old girl who was brought to the emergency department because of a 12-h history of acute onset of chest pain on the left side and shortness of breath of sudden onset, with a two-hour history of transient episodes of loss of consciousness, squinting of the eyes, and stiffening of the hands for two minutes, followed by a 10-min period of postictal state. She had no history of trauma, fever or previous respiratory symptoms. She had started her latest menstrual period two days before. There was no family history of any respiratory disease, neurologic or other medical conditions. Her past medical history, including any intellectual disability, was negative, except for a previous episode of spontaneous pneumothorax one year previously that had not been studied after treatment with chest tube thoracostomy.
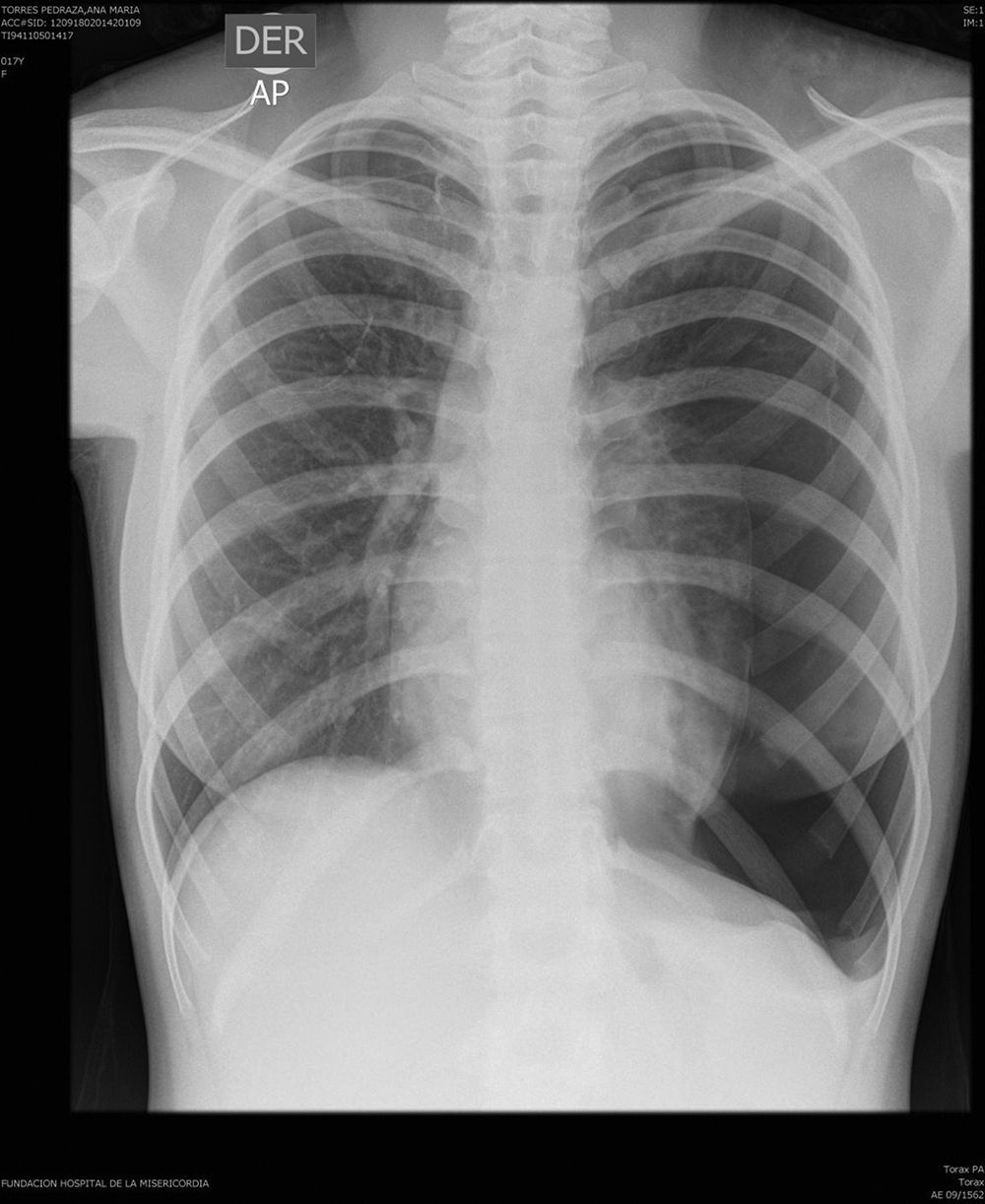
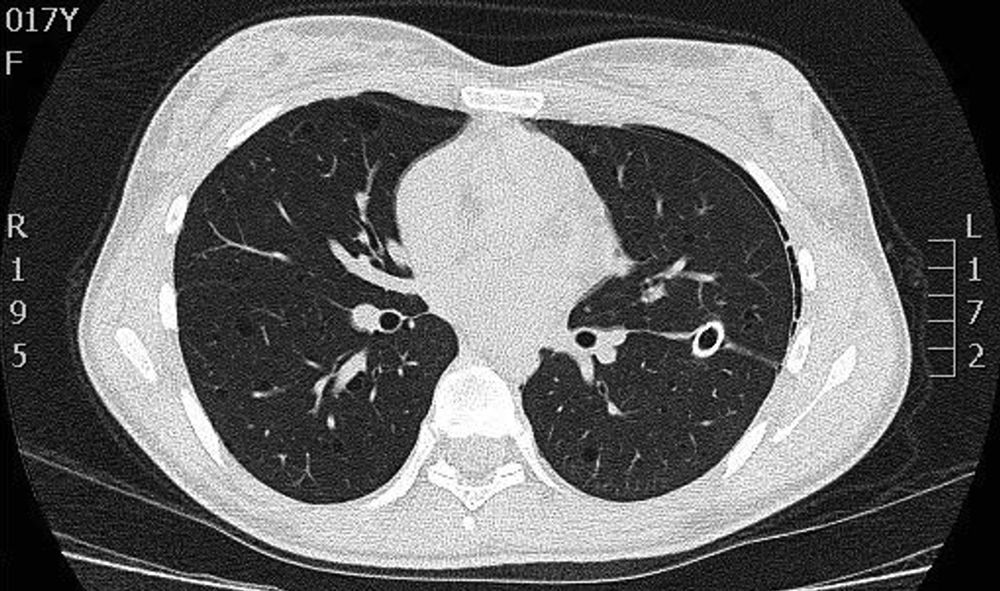
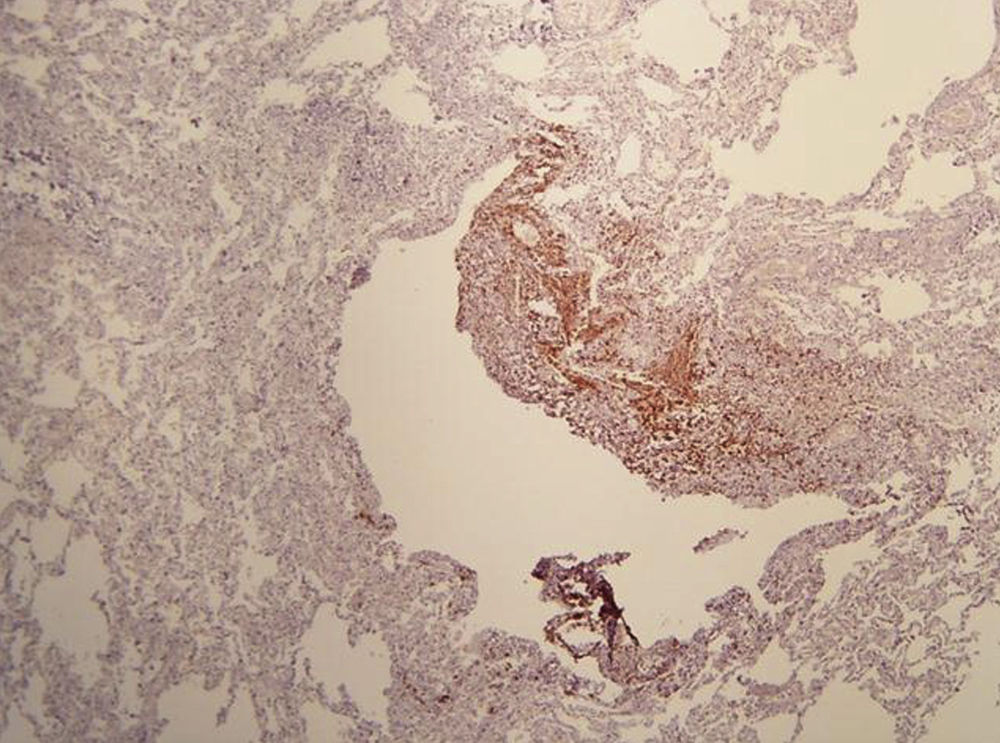
On physical examination, pulse rate was 104 beats per minute, respiratory rate was 24 breaths per minute, and pulse oximetry measurement was 94% O2 saturation in ambient air at rest, and she had decreased breath sounds on the left hemithorax. The remainder of her examination, including a thorough neurological exam, was unremarkable. Echocardiogram, ophthalmologic examination, and skin examination under ultraviolet light did not show abnormalities. A chest radiograph revealed right upper lobe cystic parenchymal lesions and a pneumothorax greater than 50% on the left side, requiring thoracostomy tube placement (Fig. 1). High-resolution computed tomography of the chest revealed multiple, well-circumscribed, round, and thin-walled cysts that were scattered in a bilateral, roughly symmetric pattern (Fig. 2). Open lung biopsy showed multiple cysts that distorted the lung parenchyma and contained proliferating bundles of “modified” smooth muscle cells in their walls, involving alveolar septa, airways, lymphatics, and blood vessels. The smooth muscle component was positive for antibodies to vimentin, actin and desmin, but it also reacted with antibodies to human melanin black 45 (HMB 45) (Fig. 3). Having established a diagnosis of LAM, the patient underwent abdominal computed tomography, which showed a rounded lesion in the upper pole of the right kidney. Three months later, she underwent nephrectomy. The piece weighed 207 grams and had a rubbery, oval mass that measured 7.5cm×6.2cm×4.5cm, well defined but unencapsulated, with yellow, ochre, and brown colours. Microscopic study showed a tripartite composition of fat, blood vessels, and smooth muscle cells in variable proportions, which reacted to the same antibodies as did the lung cysts. A diagnosis of angiomyolipoma was made. Cerebral magnetic resonance imaging with a gadolinium contrast agent revealed no abnormalities.


High-resolution computed tomography scan of the chest showing multiple well-defined, thin-walled cystic parenchymal lesions distributed diffusely throughout the lungs, and closed left thoracostomy tube with a small residual left pneumothorax, with normal cardiovascular structures.

At the present time, the patient is scheduled for the performance of a surgical pleurodesis and for administration of inhibitors of mTOR (Sirolimus).
Lymphangioleiomyomatosis (LAM) is a rare and slowly progressive multi-system disease which predominantly affects young women of childbearing age, and also occurs occasionally in males and children.1 It is characterised by an abnormal proliferation of smooth muscle-like cells (LAM cells) leading to progressive cystic destruction of the lung parenchyma, development of fluid-filled lymphatic cystic structures (lymphangioleiomyomas) in the axial lymphatics, and benign tumours, which primarily involve the kidneys (angiomyolipomas). LAM can occur in a sporadic form (S-LAM) or in association with the genetic disease TSC (TSC-LAM), an autosomal dominant syndrome of variable penetrance characterised by hamartoma formation in multiple organ systems, cerebral calcifications, seizures, mental retardation, cognitive defects, autism, and tumours of the brain, kidney, heart, retina and skin. S-LAM has an estimated prevalence of 1 in 400,000 adult females and about 34% of patients with TSC (TSC itself has a prevalence of 1 in 5800 live births).
Since the first reports of LAM in 1937, significant progress has been made in understanding the disease through the discovery of mutations in the tuberous sclerosis genes TSC1 and TSC2 as underlying causes for S-LAM and TSC-LAM. S-LAM develops due to two acquired mutations (usually in TSC2), while patients with TSC-LAM have one germline mutation (usually in TSC2) and one acquired mutation.2 For these reasons, LAM occurs frequently in association with TSC, and S-LAM is a relatively infrequent disease.
The protein products of TSC1 and TSC2 are hamartin and tuberin, respectively,3 which have an important role in the transduction of signals from cell membrane-associated receptors. Mutations in both hamartin and tuberin have been shown to inactivate the tuberin–hamartin complex, resulting in increased activity of a kinase known as the mammalian target of rapamycin (mTOR) (a central regulator of cell growth), leading to proliferation of LAM cells. The pathogenesis of LAM also comprises LAM-cell propagation through lymphatic channels, secretion of two lymphangiogenic growth factors: VEGF-D (vascular endothelial growth factor D) and VEGF-C (vascular endothelial growth factor C) by LAM cells,4 oestrogen (through interaction with signalling events in LAM cells), and altered metabolism of extracellular matrix.5,6
Clinical manifestations of LAM are pneumothorax from cyst rupture (which is often the first manifestation and tends to be recurrent), progressive dyspnoea (which is the result of airflow obstruction and cystic destruction of the lung parenchyma), and less commonly chylous pleural effusions. Other respiratory manifestations are cough, chyloptysis, and haemoptysis. Extrapulmonary manifestations are angiomyolipomas, which occur mostly in the kidneys, chylous ascites, abdominal lymphadenopathy, and large cystic lymphatic masses termed lymphangioleiomyomas. Finally, signs consistent with TSC, such as facial angiofibromas, periungual fibromas, nail ridging and the shagreen patch, can be seen in patients with TSC-LAM. S-LAM and TSC-LAM have differential characteristics. Compared to patients with TSC-LAM, patients with S-LAM have a higher frequency of lung cysts, chylous plural effusion, and abdominal lymphangioleiomyomas. Patients with TSC-LAM have a higher frequency of elevated serum levels of VEGF-D, pneumocyte hyperplasia, renal angiomyolipomas, and perivascular epithelioid cell tumour (‘PEComa’) of the uterus.
Based on pathological and clinical findings, extrapulmonary manifestations, and high-resolution computed tomography (HRCT) scans, the diagnosis of LAM can be defined as definite, probable, or possible. The diagnosis of LAM is considered definite in the presence of: (1) characteristic HRCT lung changes (>10 thin-walled, round, well-defined, air-filled cysts, 2–5mm in diameter and up to 30mm in size, distributed evenly throughout the lungs with normal lung pulmonary parenchyma), and (2) lung biopsy fitting the pathological criteria for LAM (including immunohistochemical reactivity with HMB-45), angiomyolipoma of the kidney, thoracic or abdominal chylous effusion, lymphangioleiomyoma, or lymph-node involved by LAM.7,8
Treatment is mainly supportive and includes bronchodilators, supplemental oxygen if necessary, pulmonary rehabilitation, prophylactic vaccinations, and treatment of complications (surgical or chemical pleurodesis, low-fat diet, therapeutic thoracentesis, thoracic duct ligation, and lung transplantation if appropriate). Current management options for renal angiomyolipomas include embolisation, and partial or total nephrectomy, with the former being preferred to the later. Yearly follow-ups should also be considered for these renal tumours. Hormonal therapy (mostly using progesterone) should only be used in individual cases with rapid progression of the disease. Inhibitors of mTOR (Sirolimus) and inhibitors of matrix metalloproteinase (MMPs) and angiogenesis (Doxycyline) are promising new therapeutic strategies. Given that early therapy with Sirolimus can stabilise lung function,9 and sometimes resolve pulmonary disease in LAM,10 the prompt identification of atypical early forms of LAM in young female adolescents, in cases such as our patient, can significantly impact the clinical outcome of paediatric patients with this condition.
ConclusionsLymphangioleiomyomatosis is a rare disease that only occasionally affects children. The disease should always be considered in the presence of spontaneous pneumothorax along with multiple thin-walled cystic parenchymal lesions. One should also look for characteristic features of tuberous sclerosis complex. Accurate and early diagnosis, coupled with appropriate treatment, is essential in order to minimise secondary progressive impairment of pulmonary function.
Conflict of interestThe authors have no conflict of interests to declare.
Funding sourceThis work was supported in part by the National Institute of Health (NIH) Career Development Award 1K12HL090020/NHLBI, Bethesda, Maryland, U.S.A. (GN).
Ethical disclosuresPatients’ data protectionConfidentiality of Data. The authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentRight to privacy and informed consent. The authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human subjects and animals in researchThe authors declare that no experiments were performed on humans or animals for this investigation.
We thank Mr. Charlie Barret for his editorial assistance.
