


There are two inheritance patterns, the X-linked recessive (XL) pattern and the autosomal recessive pattern. There is no information on the predominant inheritance pattern of male patients with chronic granulomatous disease (CGD) in Mexico.
ObjectiveThe aim of this study was to determine the inheritance pattern in a cohort of Mexican male patients with CGD by means of the detection of an XL status carrier among their female relatives, and to describe the frequency of discoid lupus (DL) among carriers.
MethodsWe detected the female relatives within the families of male patients with CGD, and carried out the 123 dihydrorhodamine (DHR) assay in all female participants. All carriers were questioned for current or past established DL diagnosis.
ResultsWe detected 33 families with one or more CGD male patients; we found an XL-CGD in 79% of the relatives from at least one female relative with a bimodal pattern. For the remaining seven relatives we were not able to confirm a carrier status by means of a DHR assay. Moreover, we detected one mother with CGD secondary to skewed X-chromosome inactivation. We also found 47 carriers, and only one carrier with DL among them.
ConclusionWe concluded that XL-CGD is the most frequent form of CGD in a cohort of CGD male patients in Mexico. DHR assay is a fast and practical tool to determine the CGD form in the Latin-American countries. Finally, DL frequency in Mexico is lower than that reported in the literature for other regions of the world.
Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by a defect in any of the five components of the NADPHoxidase that leads to a complete lack of, or significant decrease in, the production of microbicidal reactive oxygen metabolites. Mutations in the gp91phox gene (CYBB on Xp 21.1) produce the X-linked recessive form of the disease that affects about 70% of all CGD patients. As expected from genetics, the overwhelming majority of the X-linked patients are male. The remaining 30% of the cases are inherited in an autosomal recessive (AR-CGD) way affecting males and females equally. Mutations in genes of NCF1, NCF2, NCF4 y CYBA are responsible for the autosomal recessive pattern. Individuals with X-CGD have a more severe clinical phenotype and higher mortality than those with AR-CGD.1–4
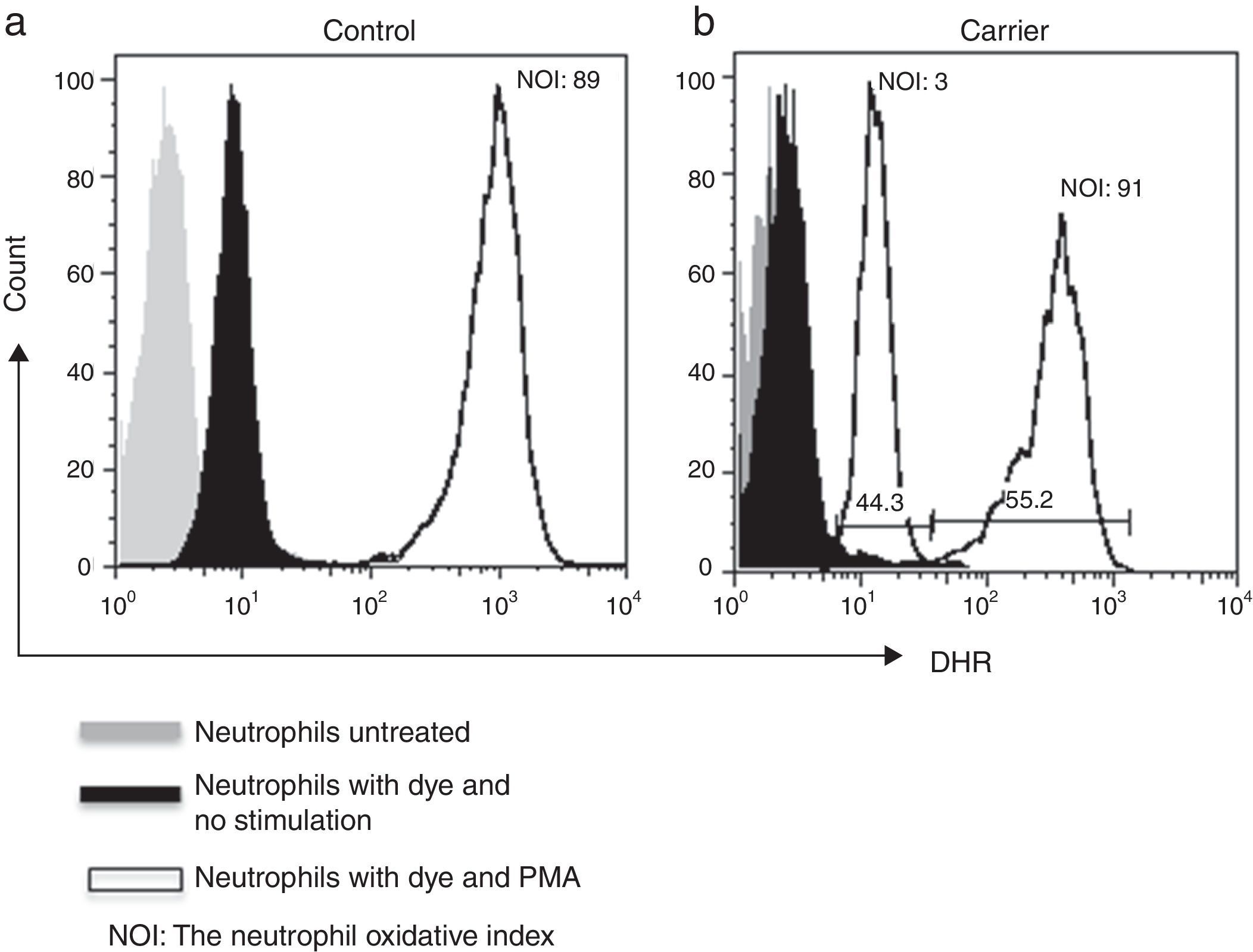
Lyonisation explains that there is a random inactivation of one X-chromosome in somatic cells during early foetus development. This results in two distinct populations of polymorphonuclear leucocytes: one with normal and the other with abnormal production of microbicidal reactive oxygen metabolites. Both populations can be detected by DHR assay, which shows a bimodal histogram for carriers.5,6
The objective of this study was to identify the inheritance pattern of the 33 Mexican males with CGD by looking for a carrier in their female relatives by means of a DHR assay. We also determined the presence, or the history, of discoid lupus in the carriers and offered them genetic counselling.
Patients and methodsPatientsThe parents with one or more male patients with CGD and without an established inheritance pattern, from eight different hospitals throughout Mexico, were invited to participate in our study. We first traced the family tree in order to identify all the female relatives on the mother's branch, and then invited them to participate in the study. The protocol was reviewed and approved by the appropriate local Ethics and Research Committees in accordance to the International Conference on Harmonization Good Clinical Practice guidelines and the Declaration of Helsinki. All participants who agreed to participate signed the informed consent and/or the informed assent. All carriers were interrogated for current or past diagnosis of discoid lupus established by a physician as disc-shaped lesions, erythematous plaques of varying size, and containing areas of follicular hyperkeratoses. Again, genetic counselling was offered to all carriers.
Three millilitres of venous blood was obtained from each participant and collected in a lithium heparin vacutainer tube. The samples were processed immediately with the objective of avoiding cell damage caused by blood drawing, shipping, mechanical irritation or temperature variation which could cause a false X inactivation of some neutrophils.
Materials and methodsThe working dilution of dihydrorhodamine 123 (DHR 123) (45μg/ml) was prepared by adding 30μl of DHR (DHR; Molecular Probes, Eugene, OR, USA) stock solution (5mg/ml) to 3.33ml of phosphate-buffered saline (PBS). In order to prepare the working dilution of phorbol-myristate-acetate (PMA) (50ng/ml), we added 10μl of PMA (Sigma Chemical, Munich, Germany) stock solution (1mg/ml) to 1ml of PBS.
Dihydrorhodamine flow cytometry assayThree 100μl samples were taken from each whole blood of possible carriers and placed in separate tubes. The tubes were labelled as stimulated,1 resting,2 and reagent blank tests.3 Twenty-five microliters of working DHR solution (final concentration 1.125μg/ml) was added to the stimulated1 and resting samples.2 All tubes were incubated at 37°C for 15min. Then 10μl of PMA solution (final concentration 100ng/ml) was added to the stimulated tubes.3 After further 20-min incubation at 37°C, all tubes were added with 1.0ml of FACS lysing solution (Becton Dickinson, Heidelberg, Germany), allowed to rest at room temperature for 20min, were then centrifuged. The supernatant was discarded and the cells were washed twice with 2ml of PBS. After the second centrifugation, the supernatant was discarded and replaced with 0.25ml of 1% paraformaldehyde.
By using the FACSAria I® (Becton Dickinson, Heidelberg, Germany) the parameters forward and side scatter were collected, as well as FL2. Twenty thousand events were acquired in the established granulocyte gate for each tube. Data analysis was performed using FlowJo 7.2.4 software (Tree Star, Inc., Ashland, OR, USA). The oxidative index (NOI) was calculated by dividing the mean fluorescence intensity of the stimulated tube by the mean fluorescence intensity of the resting tube. One woman showing a bimodal histogram pattern in the DHR assay was verified twice (Fig. 1), once the X-linked CGD carrier status had been corroborated a doctor gave genetic counselling.
Results
We detected 33 families with at least one male with CGD. We tested 90 women for DHR assay and detected an X-linked inheritance pattern in 26 (79%) relatives from the presence of at least one female relative with a bimodal pattern. In the remaining seven relatives we could not confirm a carrier status by means of the DHR assay.
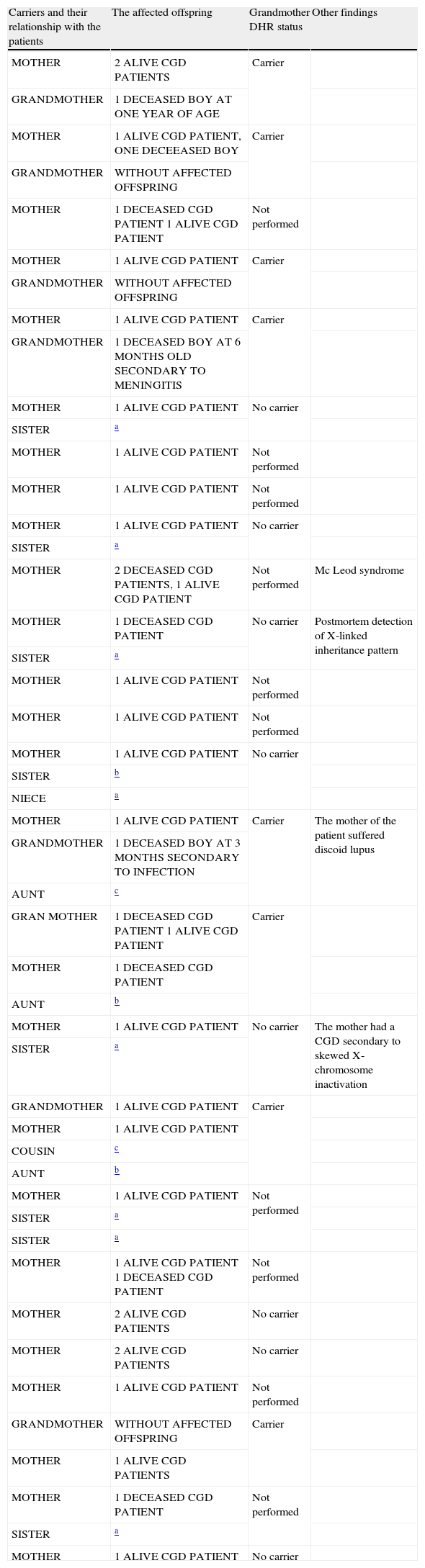
In all 26 relatives with the X-linked inheritance pattern CGD, we found a total of 47 carriers, consisting in 26 mothers, eight sisters, eight grandmothers, three aunts, one cousin, and one niece (Table 1).
47 carriers in 26 families with X-linked CGD inheritance pattern detected through 123-DHR test.
| Carriers and their relationship with the patients | The affected offspring | Grandmother DHR status | Other findings |
| MOTHER | 2 ALIVE CGD PATIENTS | Carrier | |
| GRANDMOTHER | 1 DECEASED BOY AT ONE YEAR OF AGE | ||
| MOTHER | 1 ALIVE CGD PATIENT, ONE DECEEASED BOY | Carrier | |
| GRANDMOTHER | WITHOUT AFFECTED OFFSPRING | ||
| MOTHER | 1 DECEASED CGD PATIENT 1 ALIVE CGD PATIENT | Not performed | |
| MOTHER | 1 ALIVE CGD PATIENT | Carrier | |
| GRANDMOTHER | WITHOUT AFFECTED OFFSPRING | ||
| MOTHER | 1 ALIVE CGD PATIENT | Carrier | |
| GRANDMOTHER | 1 DECEASED BOY AT 6 MONTHS OLD SECONDARY TO MENINGITIS | ||
| MOTHER | 1 ALIVE CGD PATIENT | No carrier | |
| SISTER | a | ||
| MOTHER | 1 ALIVE CGD PATIENT | Not performed | |
| MOTHER | 1 ALIVE CGD PATIENT | Not performed | |
| MOTHER | 1 ALIVE CGD PATIENT | No carrier | |
| SISTER | a | ||
| MOTHER | 2 DECEASED CGD PATIENTS, 1 ALIVE CGD PATIENT | Not performed | Mc Leod syndrome |
| MOTHER | 1 DECEASED CGD PATIENT | No carrier | Postmortem detection of X-linked inheritance pattern |
| SISTER | a | ||
| MOTHER | 1 ALIVE CGD PATIENT | Not performed | |
| MOTHER | 1 ALIVE CGD PATIENT | Not performed | |
| MOTHER | 1 ALIVE CGD PATIENT | No carrier | |
| SISTER | b | ||
| NIECE | a | ||
| MOTHER | 1 ALIVE CGD PATIENT | Carrier | The mother of the patient suffered discoid lupus |
| GRANDMOTHER | 1 DECEASED BOY AT 3 MONTHS SECONDARY TO INFECTION | ||
| AUNT | c | ||
| GRAN MOTHER | 1 DECEASED CGD PATIENT 1 ALIVE CGD PATIENT | Carrier | |
| MOTHER | 1 DECEASED CGD PATIENT | ||
| AUNT | b | ||
| MOTHER | 1 ALIVE CGD PATIENT | No carrier | The mother had a CGD secondary to skewed X-chromosome inactivation |
| SISTER | a | ||
| GRANDMOTHER | 1 ALIVE CGD PATIENT | Carrier | |
| MOTHER | 1 ALIVE CGD PATIENT | ||
| COUSIN | c | ||
| AUNT | b | ||
| MOTHER | 1 ALIVE CGD PATIENT | Not performed | |
| SISTER | a | ||
| SISTER | a | ||
| MOTHER | 1 ALIVE CGD PATIENT 1 DECEASED CGD PATIENT | Not performed | |
| MOTHER | 2 ALIVE CGD PATIENTS | No carrier | |
| MOTHER | 2 ALIVE CGD PATIENTS | No carrier | |
| MOTHER | 1 ALIVE CGD PATIENT | Not performed | |
| GRANDMOTHER | WITHOUT AFFECTED OFFSPRING | Carrier | |
| MOTHER | 1 ALIVE CGD PATIENTS | ||
| MOTHER | 1 DECEASED CGD PATIENT | Not performed | |
| SISTER | a | ||
| MOTHER | 1 ALIVE CGD PATIENT | No carrier |
Grandmothers and mothers of 26 relatives were tested for DHR assay; eight grandmothers showed a bimodal pattern and therefore were assigned with the carrier status. For the remaining eight grandmothers we did not find any carrier, even in other family members of their family other than patient's mother and sister.
As for family # 11, the mother of a dead CGD son participated; she and her daughter showed a histogram bimodal pattern.
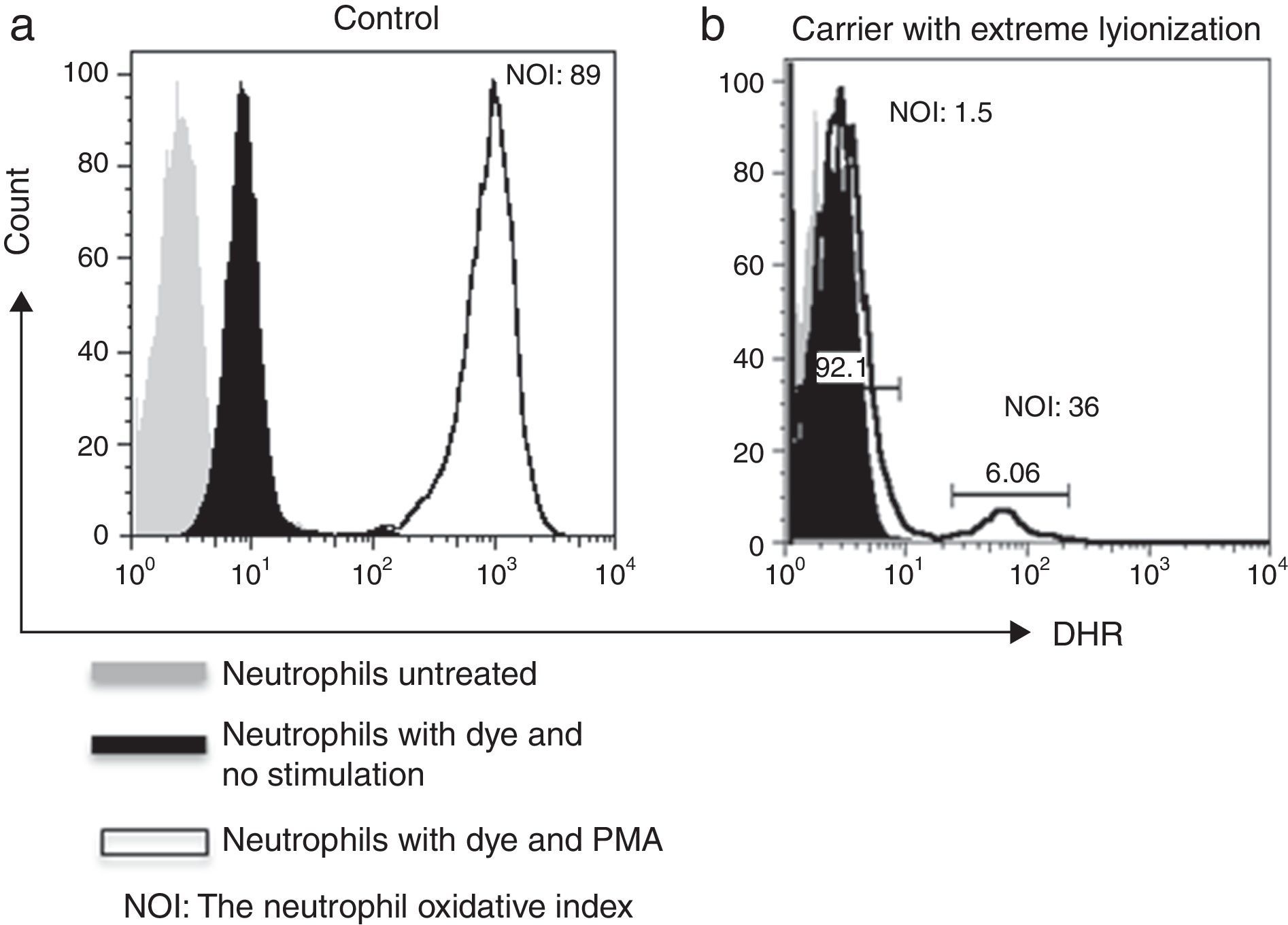
As for family # 17, we found that the mother had a CGD secondary to skewed X-chromosome inactivation. DHR assay showed only 5.3% functional neutrophils (Fig. 2). At the time of the study, she was 30 years old, she had a history of recurrent episodes of cervical suppurative lymphadenopathies and a liver abscess since she was 15 years old, but she was healthy before that age. Her daughter also resulted a carrier. Her mother and her five sisters had a normal pattern from DHR test.

None of the carriers showed current or past mycobacterial infection.
Only one of 47 carriers (2.1%) suffered from discoid lupus in the third decade of life (family # 15).
DiscussionX-linked CGD is the most prevalent inheritance pattern around the world; in a cohort of 33 Mexican families we found an X-linked pattern in 26 (79%) families; for the remaining seven families we cannot conclude X-CGD or CGD-AR from the DHR assay; because in the case of the skewed X-chromosome inactivation, all cells will yield a positive result for DHR oxidation and therefore a bimodal histogram pattern will not be seen in carriers’ DHR assay. In this case, additional examination of sisters, grandmother or nephews could reveal a bimodal histogram pattern. HUMARA analysis is necessary in order to confirm or discard lyonisation defect in the patient's mother. If the patient's mother shows no bimodal pattern, novo mutation in CYBB gene from the patient's or mother's germinal cells is another feasible explanation. The third and most likely explanation is the presence of AR-CGD in a carrier showing a normal histogram, for whom diagnosis must be confirmed by means of a molecular study.6–8
Another practical advantage of DHR assay performed on the patient's female relatives in our study was the determination of an X-linked pattern in a deceased CGD male. We retrospectively detected a carrier status in a patient's mother and sister (family # 11), and we were able to offer genetic counselling.9
As for family # 17, we found that DHR assay performed in the patient's mother showed a pattern compatible with CGD, and that the patient's sister was also a carrier. We concluded that the patient's mother had an extremely skewed X chromosome inactivation. Inactivation of the X-chromosome in carriers has given different proportions of positive and negative cells depending on which X chromosome is more likely to be inactivated, ranging from normal to pathological results.10 Thus an advantage of performing DHR assay pairwise for patients and their mothers is the detection of a skewed X chromosome inactivation. Grandmothers and aunts were normal in DHR suggesting a novo mutation in mothers’ CYBB gene; molecular study is the confirmatory test. The degree of mosaicism may vary over time in X-CGD carriers as we have shown for the mother who clinically began with recurrent and severe infections since the age of 15 years. Each carrier must be advised on that risk as part of the genetic counselling.8,11
In 16 families in which the grandmother was evaluated, we confirmed a carrier status in eight patients and did not find evidence of it in eight. It could be explained secondary to a novo mutation in mothers’ CYBB gene or to an inactivation of the X-chromosome with the mutation producing a normal result in the DHR assay. Molecular studies are the confirmatory tests.
X-linked carriers show a mosaic pattern for normal and defective neutrophils on oxidative testing by DHR, with oxidase-positive cells ranging in most cases from 20% to 80%.12 In rare instances, carriers with 10% oxidase-positive cells have normal host defence, as we have found in the grandmother from family# 15.
In a cohort of 16 X-linked CGD carriers from Netherlands, 31% of women had clinically discoid lupus. The fact that we have found a lower percentage could be associated to ethnical factors, but it could also be explained by a lack of diagnosis.13
In conclusion we detected the X-linked inheritance pattern as the most prevalent in a Mexican cohort of CGD male patients, as it has been reported in most countries around the world.
Ethical disclosuresConfidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in this study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human subjects and animals in researchThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and World Medical Association and the Helsinki Declaration.
Conflict of interestThe authors have no conflict of interest to declare.
This work was supported by CONACyT SALUD-2012-01-180910 and Fundación Mexicana para niñas y niños con inmunodeficiencias primarias A.C. Lizbeth Blancas has an SNI fellowship. Laura Berrón-Ruiz was the recipient of a postdoctoral (CVU 44813) scholarship from CONACyT, México. Andrea Morin Contreras was the recipient of a master scholarship from CONACyT (376697).
