


Auto-inflammatory syndromes are primary immunodeficiencies characterised by spontaneous and recurrent systemic inflammation episodes without an underlying infectious, neoplastic or autoimmune origin. These diseases are little known, and are therefore underdiagnosed.
One such disorder is Majeed syndrome,5 a rare autosomal recessive disease manifesting as early-onset chronic recurrent multifocal osteomyelitis (CRMO)2,7–10 with haematological alterations (dyserythropoietic microcytic anaemia), and which is sometimes also accompanied by recurrent fever or neutrophilic dermatosis. The syndrome is caused by homozygous mutations of the LPIN2 gene located on chromosome 18p.3,4
Children with Majeed syndrome5,6 suffer recurrent joint pain sometimes accompanied by fever, and with radiological findings indistinguishable from those of infectious osteomyelitis. However, the bone marrow biopsy cultures are sterile, and antibiotic treatment therefore does not lead to improvement. Analgesics and corticosteroids are partially effective. The disease evolves towards persistent joint inflammation with secondary contractures and deformities, and important functional disability.
Interleukin-1 receptor antagonists (IL-1RA) have been prescribed to treat some auto-inflammatory syndromes, although there is little experience on their use in children with Majeed syndrome.1 Two cases have been reported to date, with satisfactory results following the administration of IL-1RA1 – this supporting the idea that IL-1 dysregulation might play an important role in the pathogenesis of the syndrome.
Clinical caseA 13-year-old boy presented with a history of macrocephalia, low body height, speech and learning retardation, attention deficit disorder and alpha-thalassemia minor. From six years of age he suffered recurrent episodes of pain and spontaneous swelling of the small and medium-sized joints (ankle, dorsal region, heel and big toe of both feet, wrists and elbows) and functional impotency, with no fever, intercurrent infection or clear triggering factor.
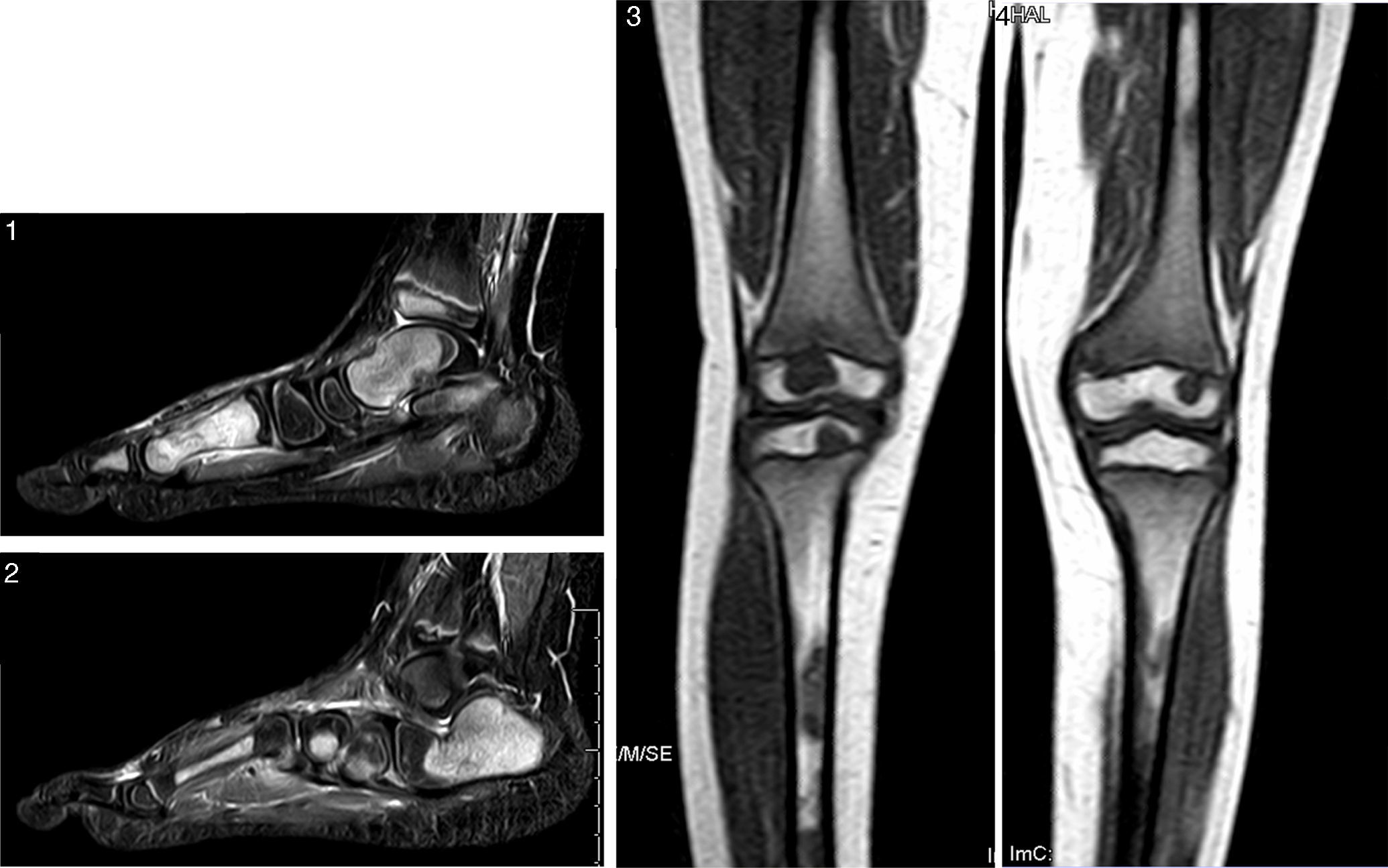
At 10 years of age he was evaluated in the Department of Paediatrics of our hospital, including a whole-body and left foot MRI scan (Figs. 1–4) and a CT-guided left calcaneal bone biopsy with bone marrow aspiration. An infectious or neoplastic origin was discarded, and chronic recurrent multifocal osteomyelitis (CRMO) was diagnosed.2,7–10 Bisphosphonates (pamidronate), nonsteroidal anti-inflammatory drugs, corticosteroids and potent opioid analgesics (Fig. 5) proved scantly effective, and the patient required frequent hospital admissions for morphic drug perfusion.
 and T1W knee and leg scan. Important replacement of the normal adipose marrow component with nodular images (hypointense in T1W and hyperintense in T2W sequencing with STIR), particularly apparent in the metacarpal region, astragalus, calcaneus, scaphoid bone, both tibias and fibulas, and both femurs – with no radiological evidence of aggressive behaviour. These findings would be consistent with proliferative/dysplastic disease, though other more infrequent disorders such as multifocal osteomyelitis or non-Langerhans cell histiocytosis (Erdheim-Chester disease) cannot be discarded.")
T2W MRI scan of the left foot with fat suppression (STIR) and T1W knee and leg scan. Important replacement of the normal adipose marrow component with nodular images (hypointense in T1W and hyperintense in T2W sequencing with STIR), particularly apparent in the metacarpal region, astragalus, calcaneus, scaphoid bone, both tibias and fibulas, and both femurs – with no radiological evidence of aggressive behaviour. These findings would be consistent with proliferative/dysplastic disease, though other more infrequent disorders such as multifocal osteomyelitis or non-Langerhans cell histiocytosis (Erdheim-Chester disease) cannot be discarded.

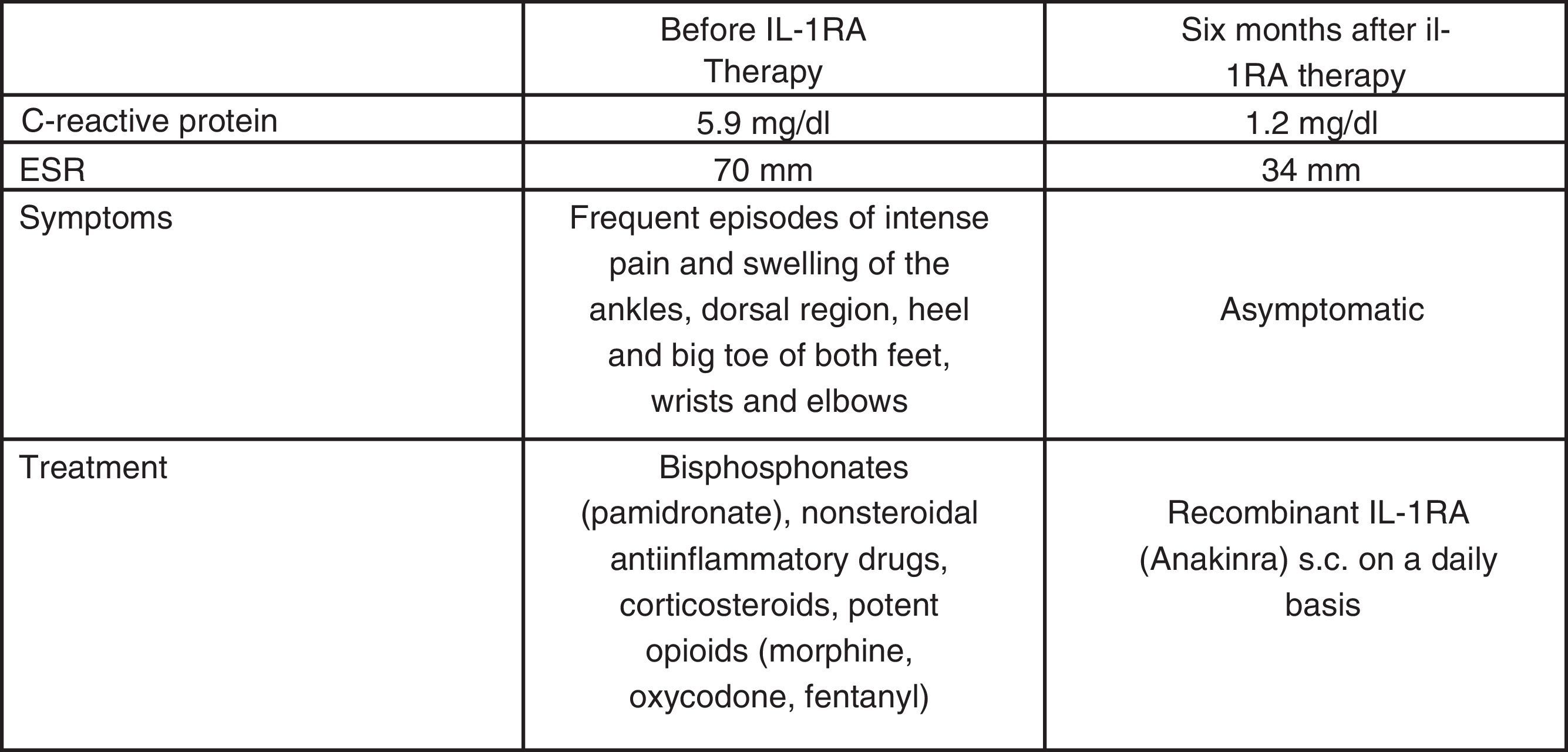
In May 2013, at 12 years of age, he was seen in the Section of Paediatric Immunology, where a clinical evaluation and thorough immunological study were carried out – the only alteration to highlight being acute phase reactant elevation during the disease flare-ups (erythrocyte sedimentation rate (ESR) up to 70mm and C-reactive protein up to 5.9mg/dl) (Fig. 5). The immunological study (immunoglobulins, complement and lymphocyte subpopulations) only evidenced a polyclonal IgG increment, and the genetic analysis revealed mutation of the LPIN2 gene in exon 17 (c.2327+1G-C).3,4
The patient was receiving third-step analgesia (WHO analgesia scale), with a progressive increase in medication requirements, increasingly frequent flare-ups (four admissions in the previous six months), and with a strong negative impact upon quality of life and family environment.
With the suspicion of Majeed syndrome,5,6 in June 2013 treatment was started with recombinant IL-1RA (Anakinra) in the Day Hospital, at a dose of 1mg/kg b.w. every 24h via the subcutaneous route1, with an excellent response allowing suspension of the analgesic treatment within two weeks. The patient has remained free of symptoms to date (Fig. 5).
CommentWe have presented the case of a boy with a variant of chronic recurrent multifocal osteomyelitis (CRMO) – Majeed syndrome5,6 – refractory to analgesic and anti-inflammatory treatment (including potent opioids), with a strong impact upon the patient's quality of life, which showed a very good response to IL-1RA treatment.1
Since the firm diagnosis was established and treatment was started, there have been no further flare-ups, and the patient has required no further hospital admissions or analgesia. He has been able to lead a normal life for a boy of his age. We thus suggest that IL-1RA treatment could be an alternative for patients with CRMO refractory to conventional therapies.1,7–10 Likewise, the results obtained support the idea (suggested by other studies) that IL-1 dysregulation might play an important role in the pathogenesis of the syndrome.1
It can be concluded that in the event of symptoms similar to those described herein, paediatricians should consider this immune disease among the possible diagnoses.
