


INTRODUCTION
T cells that use non-cytolytic mechanisms to down-regulate the immune response (suppressor T cells or Ts) are thought to play an essential role in controlling reactivity to foreign antigens and inducing tolerance to self antigens 1. Nevertheless, more than three decades since their discovery, an understanding of the mechanisms whereby suppressor cells exert their activity is still incomplete.
Phenotypically, CD8+ Ts are characterized by the lack of the CD28 co-stimulatory molecule 2. CD8+CD28 T cells are thought to arise from CD8+CD28+ T cells that have proliferated several times since they have shorter telomeres 3 and share oligoclonal expansions 4. In vitro, CD8+CD28 T cells arise from CD8+CD28+ T cells repeatedly stimulated in the presence of interleukin (IL)-2 5. In contrast, IL-4 can block this differentiation 6.
Recent evidence has been accumulating to show that CD8+CD28 T cells can inhibit T helper cell activation and proliferation in mitogen and antigen-driven responses 7-9. In vitro, antigen-specific CD8+CD28 T suppressor cells have been generated by multiple rounds of stimulation of human CD8+CD28+ T cells with APC either from an allogeneic or a xenogeneic donor 8,10. The generated cells expressed Foxp3 11, a gene related with regulatory function, and could inhibit proliferation of CD4+ T cells interacting directly with the APC used for priming 12,13.
Non-antigen specific CD8+CD28 T cells with suppressor activity have also been generated from CD8+CD28 T cells in the presence of IL-2 and IL-10 14. These suppressor cells inhibited both antigen-specific CD4 + T cell proliferation and cellular cytotoxicity by secreting cytokines such as IFN-γ, IL-6 and IL-10 15,16. Defects in this antigen-nonspecific suppression have been described in multiple sclerosis 15 and in systemic lupus erythematosus patients 17, and are primarily seen in chronic progressive situations.
Atopic allergic diseases are immune disorders caused by anomalous, Th2 cell-dominated responses to otherwise innocuous substances, such as proteins from house dust mite or grass or tree pollen 18. Many authors believe that atopy and allergic diseases are the result of inadequate or impaired inhibition of allergen-specific T helper-type responses by the regulatory T cells 19, or even a consequence of decreased frequencies of those cells in atopic individuals 20. There is a growing body of evidence which suggests that CD8+ T cells play an important part in regulating the IgE response to non-replicating antigens 21,22. Nonetheless, the presence of CD8+ suppressor T cells in human atopic patients is not yet confirmed.
MATERIALS AND METHODS
Subjects
Peripheral blood was obtained from 30 non atopic and 32 atopic (with allergic rhinitis) adult volunteers (Caucasian, non-smokers), matched for age and gender. Atopic volunteers were recruited from the allergy clinic of the Cova da Beira Hospital and non atopic volunteers were recruited among the staff of the Hospital and the University.
Atopy was assessed by positive skin prick tests and specific IgE levels to Dermatophagoides pteronyssinus (Der p). Volunteers who received immunotherapy or were on systemic medication were excluded. Pregnant or breastfeeding women and all volunteers with disease affecting the immune system were also excluded. Informed written consent was signed by all the volunteers. The study was approved by the Ethics Committee of Centro Hospitalar Cova da Beira.
Monoclonal antibodies
The following monoclonal antibodies (mAbs) were used: anti-CD3 mAb conjugated with APC was purchased from Pharmingen (San Diego, CA, USA), anti-CD8 mAb conjugated with PerCp was purchased from Becton Dickinson (San José, CA, USA), anti-CD28 mAb conjugated with FITC was purchased from Pharmingen, anti-TCRαβ, -TCRγδ, -CD25, -CTLA-4, -HLA-DR, -CD56, -CD94, -CD158a, -CD161, -NKB1 mAb conjugated with PE were all purchased from Pharmingen, anti-Vα24 mAb conjugated with PE was purchased from Serotec (Oxford, UK). Isotype-matched, PE-conjugated control mAbs IgG1, IgG2a, and IgM were purchased from Pharmingen.
Blood preparation and antibody staining
Freshly collected peripheral blood mononuclear cells (PBMC) were stained either from whole blood after lysis of the erythrocytes (10 mM Tris, 0.15 M NH4Cl, pH = 7.4). Staining was performed at 4 °C for 30 min in staining solution (PBS, 0.2 % bovine serum albumin (BSA), 0.1 % NaN3) in round-bottomed microtiter plates (Greiner, Nürtingen, Germany) with ≈ 0.5 × 106 cells/well. After staining, the cells were washed, resuspended in 500 μl PBS and acquired in a FACSCalibur Flow cytometer (Becton Dickinson).
Flow cytometry analysis
Data were collected on 20,000 cells/sample using FACSCalibur flow cytometer equipped with an argon ion laser and a red diode laser, for quantification of CD28+ and CD28 T cells. Anti-CD8, -CD3, and -CD28 monoclonal mAb were used to define the CD3+CD8+CD28+ and CD28 T cell populations. T lymphocytes were gated for analysis based on light scattering properties and on CD3 staining. Positively and negatively stained populations were calculated by quadrant dot plot analysis determined by isotype controls.
For phenotypic analysis of the defined subpopulations, 30,000 events were collected per sample. Fluorescence dot plots and histograms were analysed using cytological software (Cell Quest Pro, Becton Dickinson). Phenotypic analysis of the TCRαβ and TCRγδ was also performed on the isolated fractions of CD3+CD8+CD28+ and CD28 T cells.
Cell isolation
Peripheral blood mononuclear cells (PBMC) from the volunteers were separated from peripheral blood (120 ml) by centrifugation over Lymphoprep. Lymphocytes were further purified by allowing the cells to adhere to plastic for 1 h at 37 °C, 5 % CO2. CD8+ T cells were isolated from the supernatant by negative selection using magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany). To separate CD28+ and CD28 T cells from CD8+ cell suspensions, cells were incubated with CD28-FITC mAb (Pharmingen) and then with anti-FITC microbeads (Miltenyi Biotec). Purity of the fractions was greater than 85 % as evaluated by flow cytometry analysis. The adherent fraction was collected as well and used as APC in cell cultures after mitomycin treatment.
Cell Cultures
Responder cells were cultured at a concentration of 0.5 × 106 cells/ml for 6 days in RMPI 1640 complete medium [RPMI 1640, 2 mM L-glutamine, 1 % antibiotic/antimicotic (Sigma-Aldrich, St Louis, MO, USA), and 10 % foetal calf serum (Biochrom, Berlin, Germany)] in 96-well flat-bottomed plates (Nunc, Roskilde, Denmark) with or without APC and 10 mg/ml purified Der p extract (a kind gift from Dr. Joost Van Neerven, The Netherlands). For positive control, cells were incubated with 0.5 μg/ml OKT3 (eBiosciences, San Diego USA) or with 5 μg PHA (Sigma-Aldrich) for 3 days in the same conditions as for Der p. Fourteen hours before the end of the culture [ 3H] thymidine was added to the wells (1 μCi/well). [ 3H] thymidine (Amersham Biosciences, Uppsala, Sweden) incorporation was determined by scintillation counts in a TopCount (Perkin Elmer, MA, USA) and results were expressed as counts per minute (cpm). Mean cpm of the triplicate cultures and standard error of the mean were calculated.
Culture conditions were as follows: CD8+CD28+ cells alone, CD8+CD28 cells alone, CD8+CD28+ cells + APC + Der p, and CD8+CD28 cells + APC + Der p.
Cell co-cultures
PBMC (0.5 × 106 cells/ml) were incubated in different proportions (1:1, 2:1, 4:1) with freshly isolated CD8+CD28+ and CD8+CD28 T cells for 6 days in 96-well flat-bottomed plates with and without 10 μg/ml Der p extract. Tritiated thymidine (1 μCi/ well) was added 14 hours prior the end of the culture. Cells were harvested on fiber filters, and incorporated thymidine was determined by scintillation counting.
Cytokine production
For soluble cytokine detection in the culture supernatants, the human Th1/Th2 cytokine bead array kit was used, according to the manufacturer's instructions (Becton Dickinson).
Statistical analysis
Statistical analysis of the results was performed using Minitab 14 statistical Software. Mann Whitney U test was used to assess differences between atopic and non atopic volunteers. Wilcoxon signed rank test was used to assess differences within the same population. A p value of less than 0.05 was considered significant.
RESULTS
First, we evaluated the frequency of CD8+CD28 T cells in whole blood from 22 atopic and 18 non atopic volunteers (age 22-42) by flow cytometry. We observed that the relative percentages were not significantly different (p = 0.80), with 41 ± 14.3 and 40 ± 13.4 % of CD8+CD28 T cells in atopic and non atopic volunteers respectively. CD28 T cells were larger, morphologically more complex and expressed CD3 with higher mean fluorescence intensity (MFI) than CD28+ T cells (1048 vs 978).
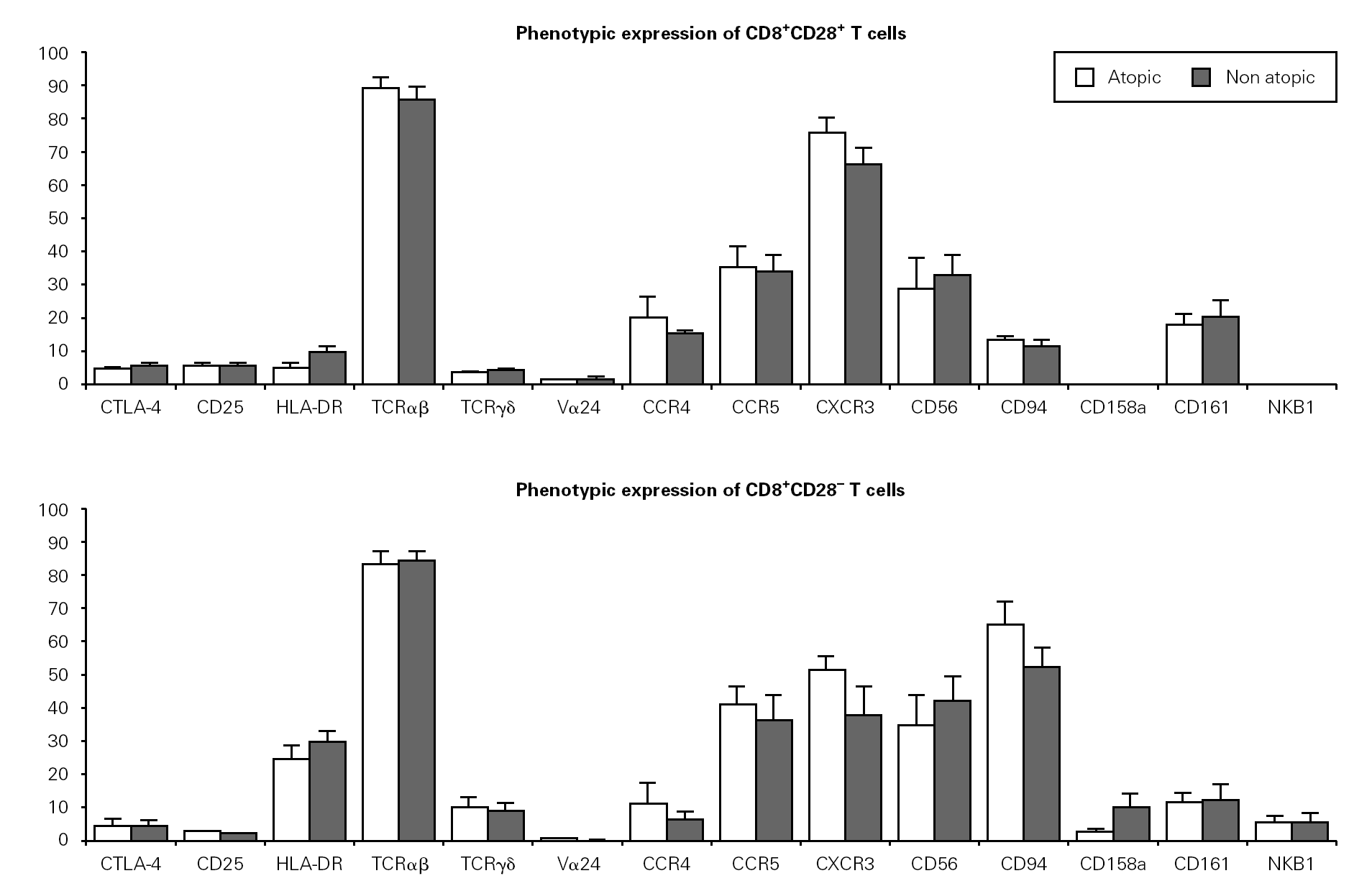
We then evaluated the expression of several phenotypic markers on CD3+CD8+CD28+ and CD28 PBMC subpopulations in whole blood from ten atopic and ten non atopic volunteers (fig. 1). Phenotypic expression was similar in both of these groups. However, taking all the data together, more CD8+CD28+ T cells than CD8+CD28 T cells expressed CD25 (5 % vs. 1.7 %) and CTLA-4 (5.5 % vs. 4 %). The reverse was true for HLA-DR expression (7.7 % vs. 29.7 %).
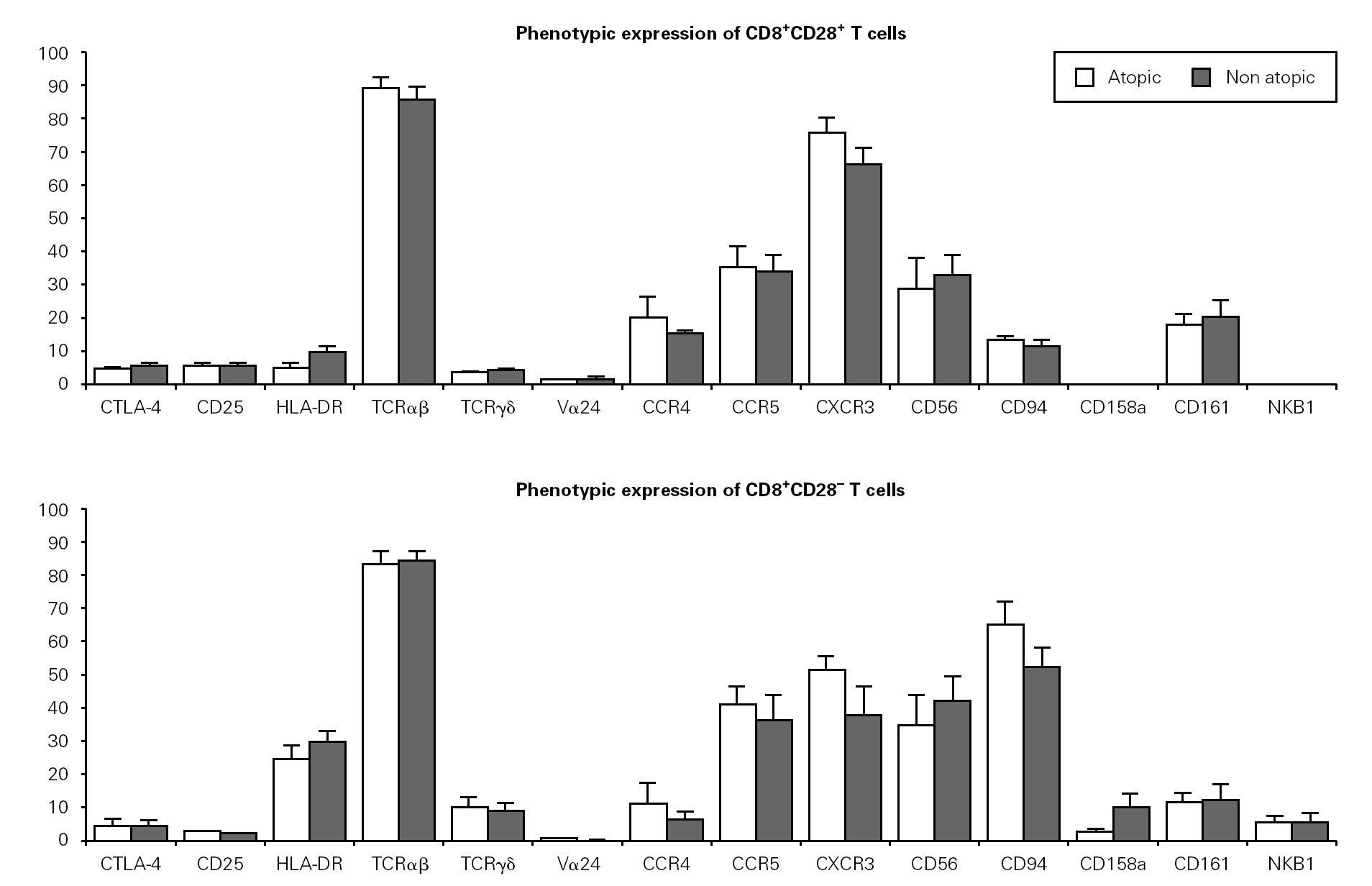
Figure 1.--Phenotypic expression on CD8+CD28+ and CD28 T cells. Freshly collected whole blood from 20 volunteers (10 atopic and 10 non atopic) was stained with CD28-FITC, CD8-PerCP, CD3-APC and PE-conjugated antibodies against one of the markers under study. Thirty thousand events were acquired in a FACSCalibur and analysed using the CellQuest Pro program. Statistical values shown (mean and s.e.m.) were obtained using the Minitab 14 program.
Next, we examined the expression of TCRαβ, TCRγδ and TCRVα24 in CD8+CD28+ and CD28 T cells. As expected, the majority of CD8+ T cells expressed a TCRαβ (87 % of CD8+CD28+ and 83 % of CD8+CD28 T cells). Interestingly, when the expression of TCRγδ was examined, we found a higher percentage of CD28 T cells than CD28+ T cells expressing that receptor (11 % vs. 4 %). TCRαβ and TCRγδ expression was also studied in the magnetically isolated CD28+ and CD28 cells fractions and the results obtained were not significantly different (data not shown).
Interestingly, the relative frequency of all the phenotypic markers related to NK cells studied (CD56, CD94, CD158a, and NKB1) was higher on CD28 T cells than on CD28+ cells. CD94 expression was considerably higher on CD8+CD28 T cells when compared to CD8+CD28+ T cells (63 % vs. 11 %). Expression of CD161 was low (17 % vs. 14 % for CD28+ and CD28, respectively) and that of CD158a and NKB1 was very low (under 10 % for both populations). Expression of chemokine receptors was also different, with the CXCR3 relative percentage being higher on CD28+ than on CD28 cells (69.6 % vs. 49.1 %). However, no difference was observed in terms of CCR5 expression between these two cell subsets. On the other hand, a higher percentage of CD28+ than CD28 T cells expressed the CCR4 chemokine receptor (21.3 % vs. 18.4 %).
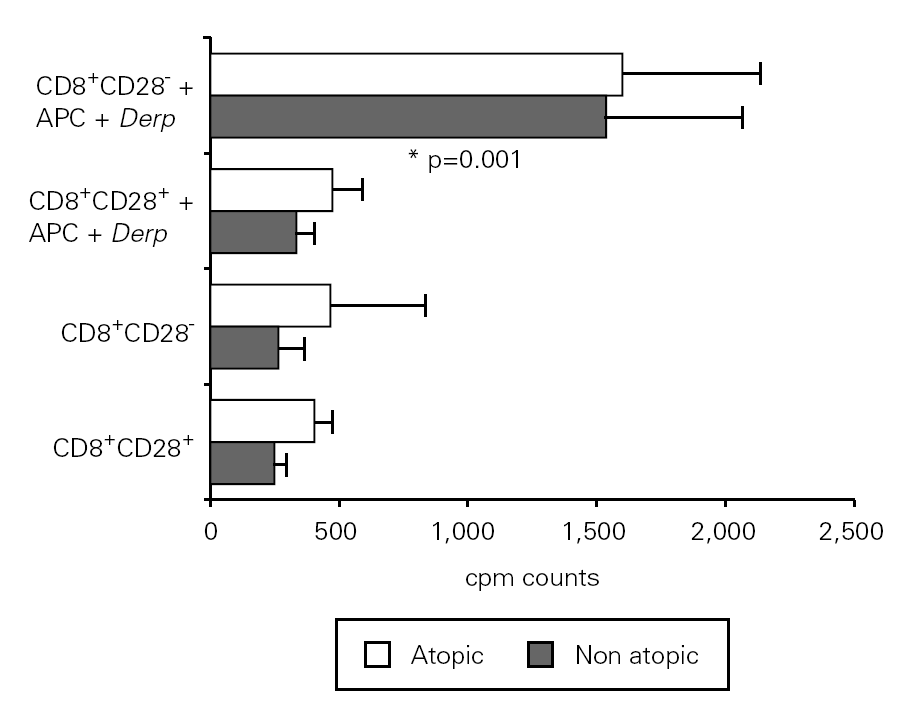
In order to assess the proliferative response to allergen of CD8+CD28+ and CD28 T cells, we set up cultures of isolated fractions with and without APC and Der p. As shown in figure 2, contrary to what occurred with unstimulated cells, CD8+ CD28 T cells proliferated when stimulated with Der p, displaying a significantly higher level (p = 0.001) of proliferation as compared with CD8+CD28+ T cells in atopic and non atopic individuals. However, the proliferation level was not significantly different between atopic and non atopic volunteers. We also stimulated some fractions with OKT3 and, in this case, CD8+ CD28 T cells proliferated less significantly (p = 0.003) than CD8+CD28+ T cells (data not shown).
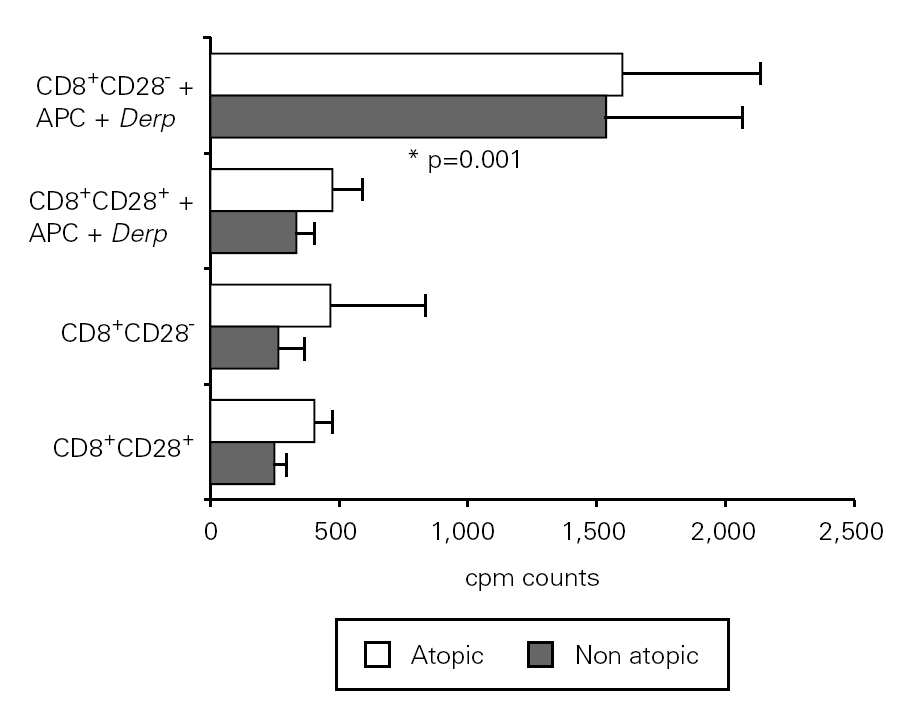
Figure 2.--CD8+CD28 T cells proliferate more than CD8+CD28+ T cells when stimulated with Der p. Isolated CD8+CD28 and CD8+CD28+ T cells from 8 atopic and 10 non atopic volunteers were incubated for 6 days in 96-well flat-bottomed plates with and without 10 μg/ml Der p extract. Tritiated thymidine (1 μCi/well) was added 14 h prior to the end of the culture. Cells were harvested on fiber filters, and incorporated thymidine was determined by scintillation counting. Results show thymidine incorporation (cpm, mean ± s.e.m.). Wilcoxon signed ranked test was used for comparison between conditions.
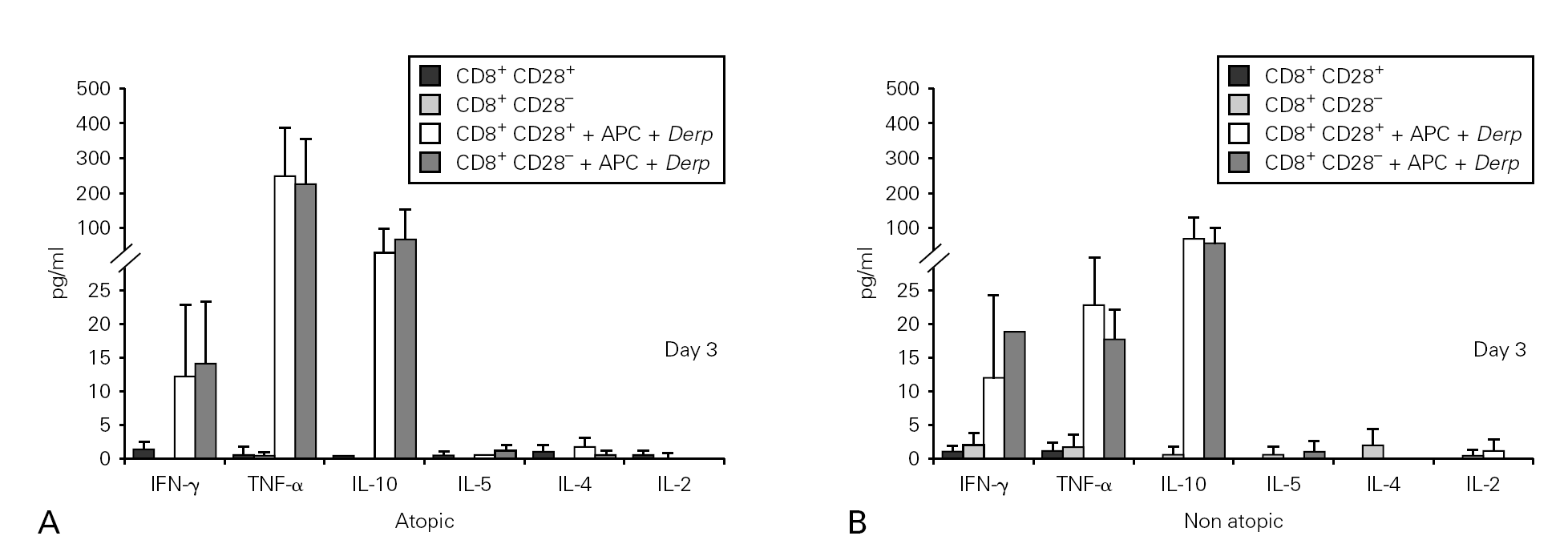
Cytokine synthesis was studied using the cytometric bead array (CBA) in culture supernatants collected on day 3. None of the cultures synthesized IL-5, IL-4 or IL-2, but all the Der p -stimulated cultures synthesised IFN-γ, TNF-α and IL-10. However, we did not observe statistically significant differences either between the cultures with CD8+CD28+ and CD28 T cells or between atopic and non atopic individuals (fig. 3).
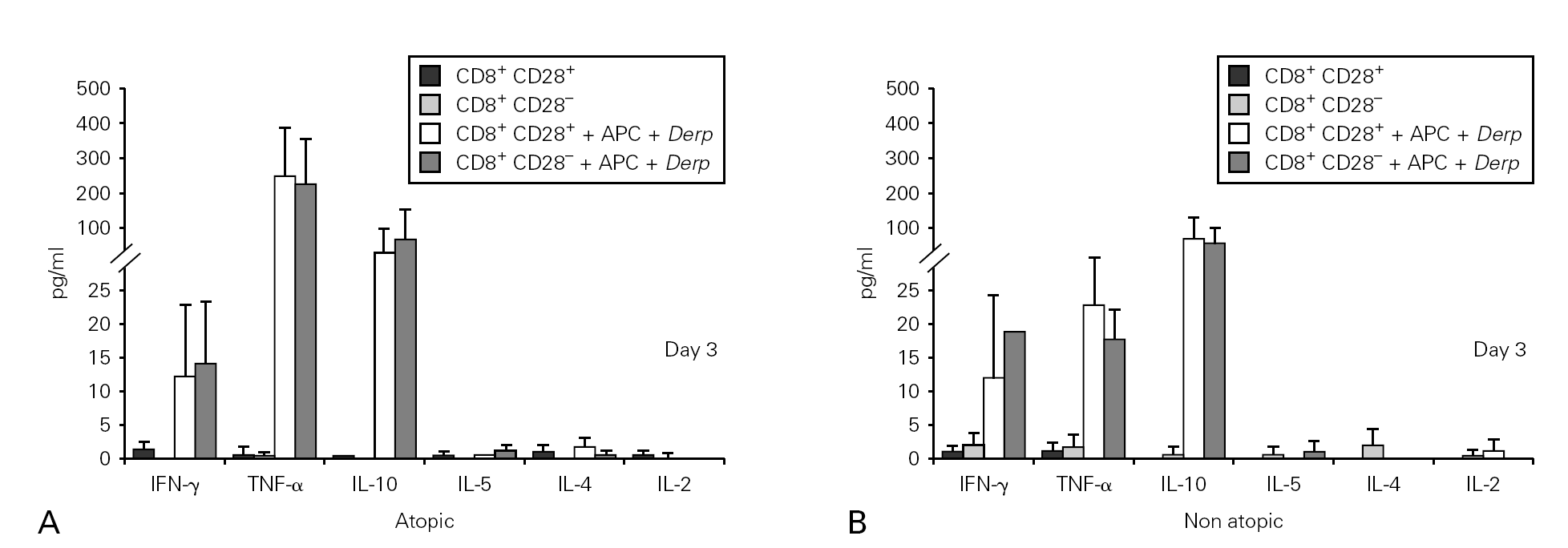
Figure 3.--Cytokine synthesis is similar for both isolated populations. Isolated CD8+CD28 and CD8+CD28+ T cells from 4 atopic and 3 non atopic volunteers were incubated in 96-well flat-bottomed plates with and without 10 μg/ml Der p extract. On day 3, culture supernatants were collected and cytokine synthesis (IFN-γ, TNF-α, IL-10, IL-5, IL-4, IL-2) was evaluated by cytometric bead array, following the manufacturer's protocol. Values shown are mean ± sem. Mann-Whiney U test was used for comparison between atopic and non atopic and Wilcoxon signed rank test was used for comparison between conditions.
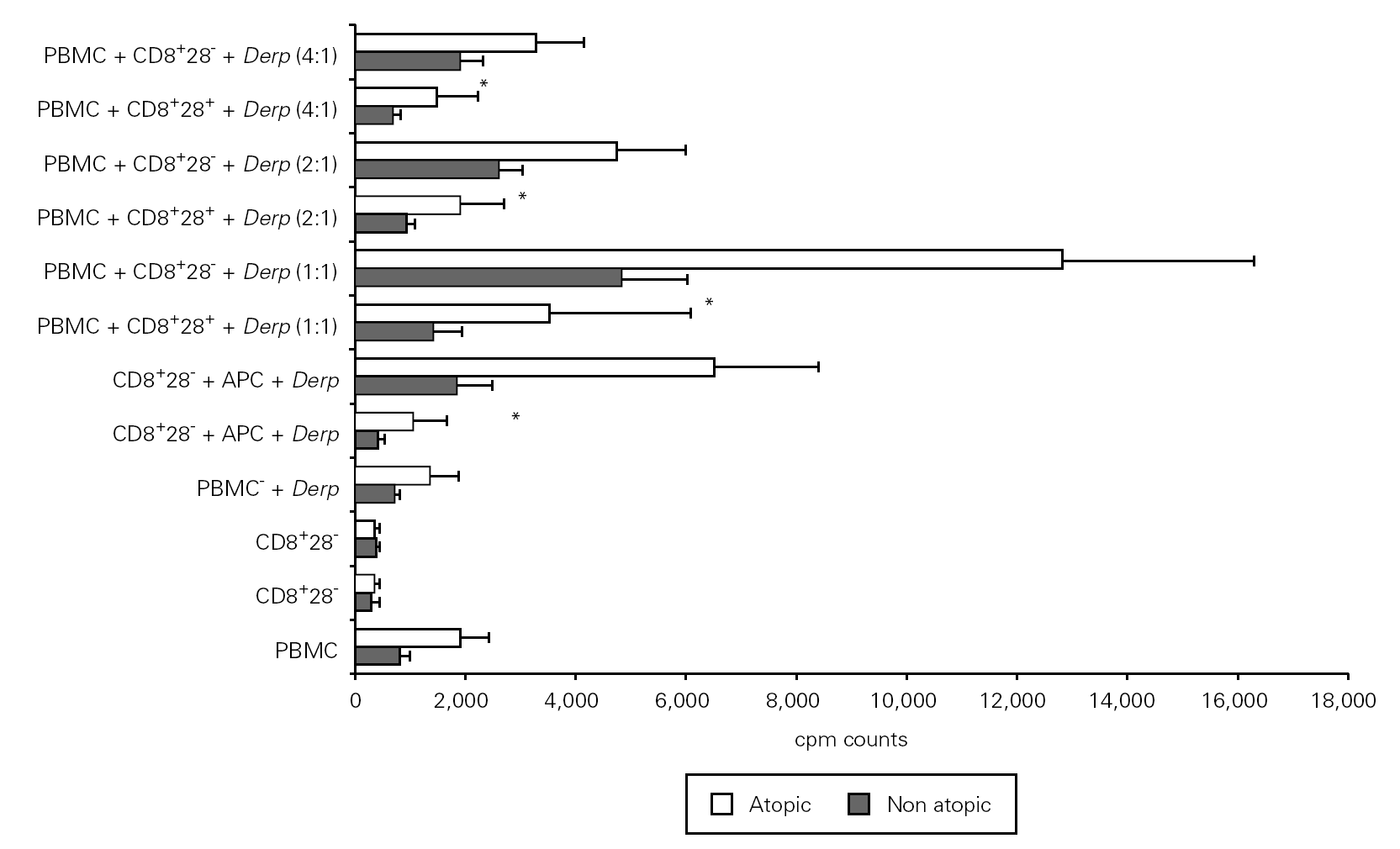
In order to study the potential suppressor properties of the isolated subpopulations, we performed co-cultures with PBMC at different ratios. None of the isolated subpopulations was able to suppress the allergen-specific PBMC proliferation, since we observed an increase in cpm counts in co-cultures (fig. 4).
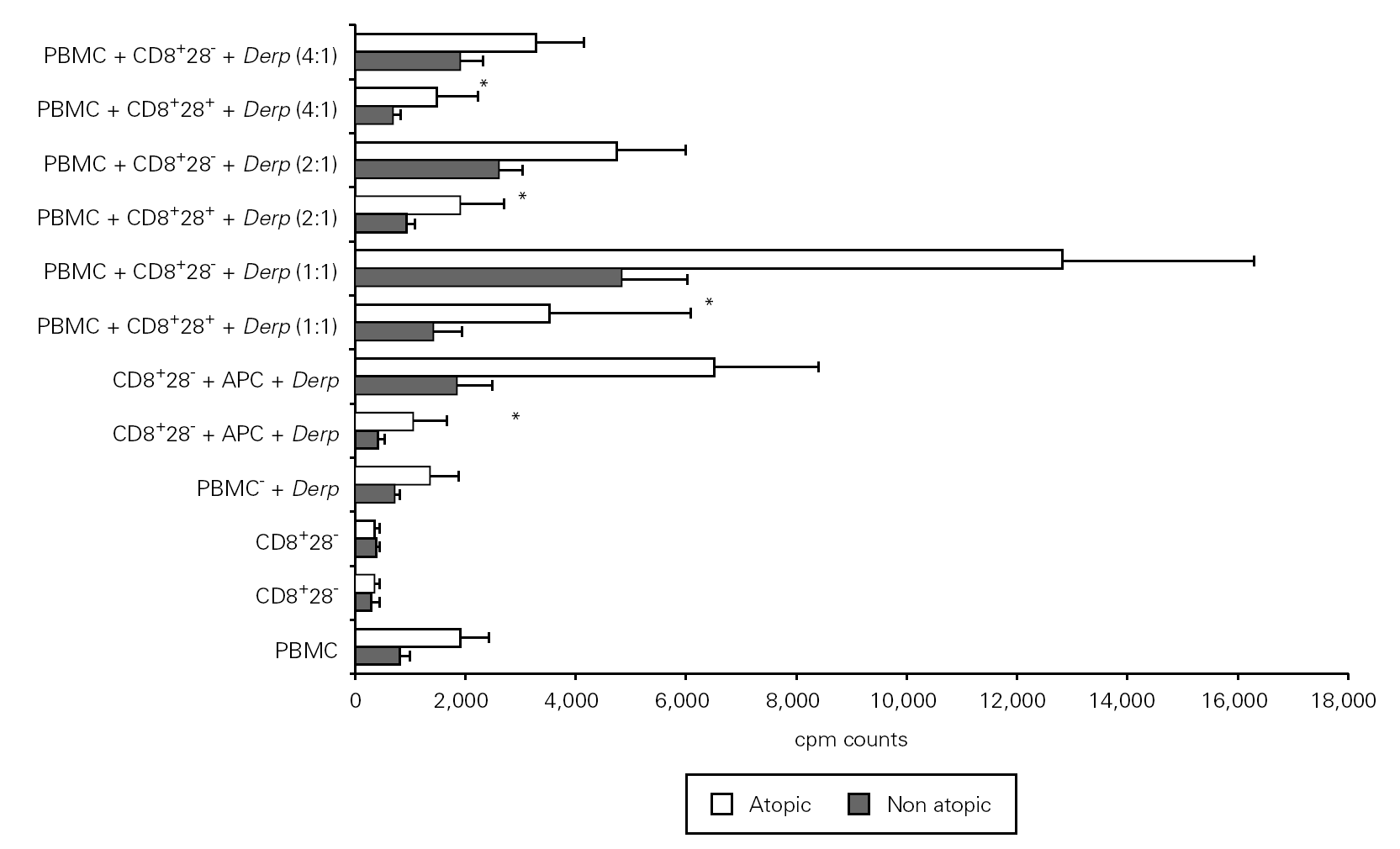
Figure 4.--CD8+CD28 co-cultures with PBMC proliferate more than CD8+CD28+ co-cultures. Isolated CD8+CD28 and CD8+CD28+ T cells from 8 atopic and 6 non atopic volunteers were incubated in different proportions with PBMC for 6 days in 96-well flat-bottomed plates with and without 10 μg/ml Der p extract. Tritiated thymidine (1 μCi/well) was added 14 h prior to the end of the culture. Cells were harvested on fiber filters, and incorporated thymidine was determined by scintillation counting. Results show thymidine incorporation (cpm, mean ± s.e.m.). Wilcoxon signed ranked test was used for comparison between conditions.
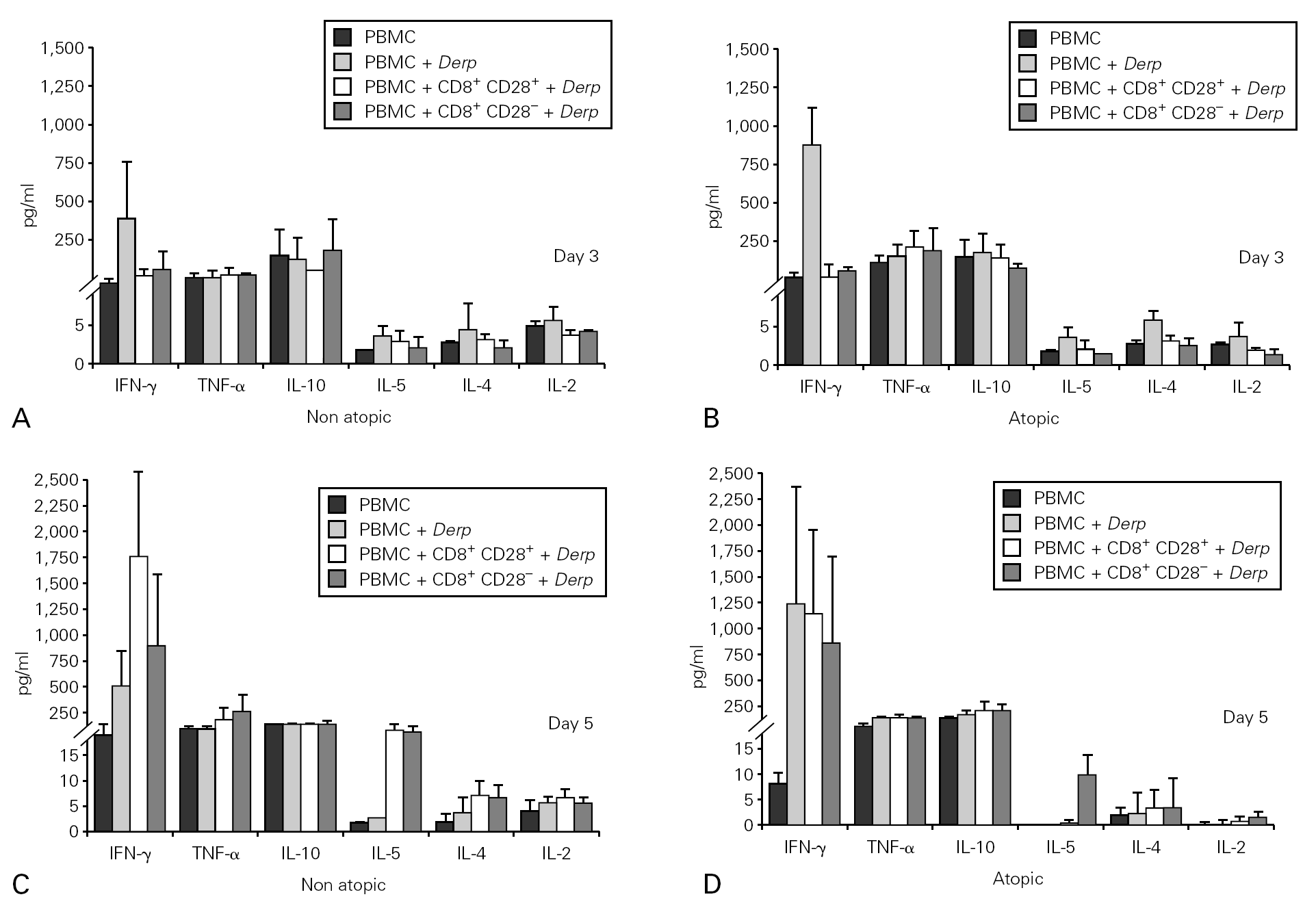
Cytokine synthesis was also evaluated in the co-culture supernatants on day 3 and on day 5, by CBA. Synthesis of IL-5, IL-4 and IL-2 was very low in all the cultures. When cytokine synthesis in the co-cultures was compared between days 3 and 5, only IFN-γ levels increased. There were no significant differences in cytokine synthesis either between cultures from atopic and non atopic or between co-cultures with CD8+CD28 or CD8+CD28+ T cells (fig. 5).
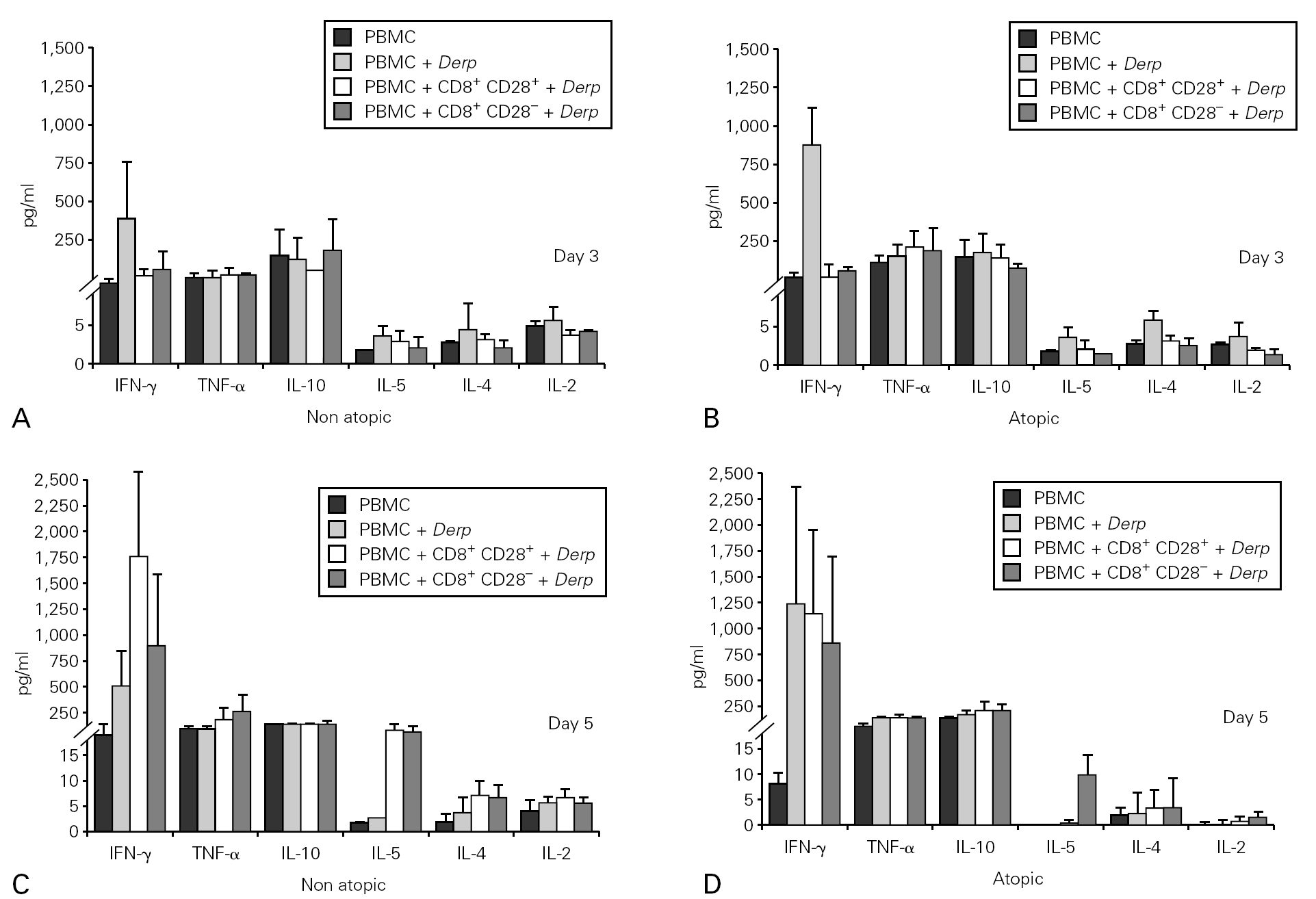
Figure 5.--Cytokine synthesis is similar in all the co-cultures stimulated with Der p. Isolated CD8+CD28 and CD8+CD28+ T cells from 4 atopic and 3 non atopic volunteers were incubated in a proportion of 1:1 with PBMC in 96-well flat-bottomed plates with and without 10 μg/ml Der p extract. On day 3 and day5, culture supernatants were collected and cytokine synthesis (IFN-γ, TNF-α, IL-10, IL-5, IL-4, IL-2) was evaluated by cytometric bead array, following the manufacturer's protocol. Values shown are mean ± sem. Mann-Whiney U test was used for comparison between atopic and non atopic volunteers and Wilcoxon signed rank test was used for comparison between conditions.
DISCUSSION
This is the first study on suppressor function of CD8+CD28 T cells in allergy. Previous studies on the regulation of atopic allergy mainly included natural T regulatory cells (CD4+CD25+) and showed inconclusive results 23,24. We believe that this study can enhance the knowledge and shed some light on the functional properties of the CD8+CD28 T cells and their implication in the development of atopy.
We observed that CD8+CD28 T cells are not phenotypically different between atopic and non atopic individuals. This may imply that atopy is not associated with a specific CD8+ T cell phenotype. However, it is important to mention that our data on phenotypic differences between CD28+ and CD28 subpopulations is in line with other reports in health and disease 25,26. Moreover, the increased expression of the NK cell related receptors in CD8+CD28 T cells, in a context where co-stimulation is not present, may be important towards limiting T cell cytolytic responses, and act as a form of "regulation".
Classical antigen-presentation studies showed that MHC class I molecules present peptides derived from proteins synthesized within the cell, whereas MHC class II molecules present exogenous proteins captured from the environment. Emerging evidence indicates, however, that dendritic cells have a specialized capacity to process exogenous antigens into the MHC class I pathway 27. According to our expectations, unstimulated cells did not proliferate. Interestingly enough, CD8+CD28 but not CD8+CD28+ T cells proliferated in response to Der p, in the presence of APCs. This fact suggests that CD28 T cell proliferation is not impaired in spite of the absence of CD28 co-stimulation 28 and that CD8+CD28 T cells respond to common aeroallergens. This results should be considered with caution as the two subsets of CD8+ T cells studied here are oligoclonal 4 with respect to their TCR, and the use of this allergen might stimulate an insignificant minority of clones and lead to inconclusive results. For this reason, we performed cell cultures for 3 days with OKT3 to confirm whether multi-clonal cellular responses were different in both subpopulations. Results show a proliferation impairment in the CD8+CD28 T cells which corroborates previous results 29,25. This implies that our results must be confirmed by performing the same studies with another allergen.
When cytokine production was analysed, no detectable IL-5, IL-4 or IL-2 production was induced by the allergen in CD28+ or CD28 T cells. On the other hand, IFN-γ, TNF-α and IL-10 were synthesized at similar levels by both isolated populations. Other investigators mention that a subpopulation of suppressor CD8+CD28 T cells synthesises IL-10, thus mediating the suppressive effect 14. Moreover, Seneviratne et al. linked low levels of IL-10 with severe atopic disease 30. Since both our subsets produced IL-10, we performed the co-culture studies in order to evaluate the "regulatory" capacity of these cells. The production of cytokines varied between donors, but followed similar patterns. The failure to detect the other cytokines (IL-5, IL-4, IL-2) in the supernatants does not necessarily imply that they are not synthesized but may also suggest that they are immediately used as autocrine factors.
Finally, we performed co-culture studies and observed that freshly isolated CD8+CD28+ or CD8+CD28 T cells did not show suppressive properties, and were not able to inhibit allergen-driven PBMC proliferation or cytokine synthesis. These results are in line with previous reports namely by Suciu-Foca and Filaci groups 31 where suppressor CD8+CD28 T cells were only generated after multiple rounds of stimulation of PBMCs with allogeneic 13, xenogeneic 8 or antigen-pulsed 32 autologous APC.
However the presence of CD8+CD28 suppressor T cells in vivo was observed in transplanted patients without rejection 33. These facts may imply a need for an elevated and sustained contact with the antigen (hence the multiple rounds of stimulation) in order to generate antigen-specific suppressor cells. It would be interesting to further study suppressive properties of CD8+CD28 T cells, by using allergen-specific T cell lines developed from atopic and non atopic volunteers.
In summary, in the present study we have shown that atopy is not associated with altered relative percentages or specific phenotypes in CD8+CD28+ or CD28 human T cells. Freshly immunomagnetically isolated CD8+CD28+ or CD28 human T cells have distinct phenotypes and, although sharing similar cytokine production patterns, they proliferate at different levels to common stimuli. Both subpopulations show similar proliferation capacity in atopic and non atopic individuals and do not have any suppressor capacity.
ACNOWLEDGEMENTS
The authors would like to thank all the volunteers without whom this study would have been impossible, and Dr. Joost Van Neerven for kindly providing the Der p extract.
O.L. is the recipient of a fellowship from the Portuguese Foundation for Science and Technology (FCT) (BD16448/2004).
Correspondence:
L. Taborda-Barata
Department of Medical Sciences
Faculty of Health Sciences
University of Beira Interior
Avenida Marques d'Avila e Bolama
6200-001 Covilhã. Portugal
Tel: + 351 275319881
Fax: + 351 275319883
E-mail: tabordabarata@ubi.pt
