


The aim was to describe the clinical manifestations, complications and long-term outcome of a cohort of Iranian patients with primary immune deficiency (PID).
MethodWe retrospectively studied the demographic, clinical and immunological characteristics of the PID patients in a single tertiary centre, from January 1989 to July 2014. The patients were classified according to the International Union of Immunological Societies Expert Committee on PID.
Results98 patients were diagnosed with and followed-up for 15 disorders. The mean age at onset and diagnosis and the diagnostic delay were 8±10, 14.2±13.1 and 6.1±7 years, respectively. Parental consanguinity rate was 57%. Predominantly Antibody Deficiency was the most common diagnosis (n=63), followed by congenital defects of phagocytes (n=16), combined immunodeficiencies (n=12), well defined syndromes (n=4) and defects in innate immunity (n=3). Recurrent sinopulmonary infection was the most common presentation. Active infections were treated appropriately, in addition to prophylactic therapy with IVIG and antimicrobials. Not all the patients were compliant with prophylactic regimens due to cost and unavailability. One SCID patient underwent successful bone marrow transplantation. The total mortality rate was 19% during the follow-up period (7.8±7.6 years). The mean age of living patients at the time of study was 23±11.7 years.
ConclusionsPhysicians awareness of PID has been rising dramatically in Iran, ensuring an increasing number of patients being diagnosed and treated. More effective treatment services, including health insurance coverage and drug availability are needed to improve the outcome of PID patients.
Primary immunodeficiency disorders (PID) are rare inherited diseases of the immune system that present with heterogeneous infectious and autoimmune manifestations and higher incidence of malignancies.1–6 The International Union of Immunological Societies (IUIS) Expert Committee classified PID in nine main groups.7 Overall, the prevalence of PID is estimated to be one in 1200 American people and appears to be underdiagnosed.2 The aim of this study was to summarise the clinical manifestations and long-term outcome of 98 Iranian PID patients in a single tertiary centre.
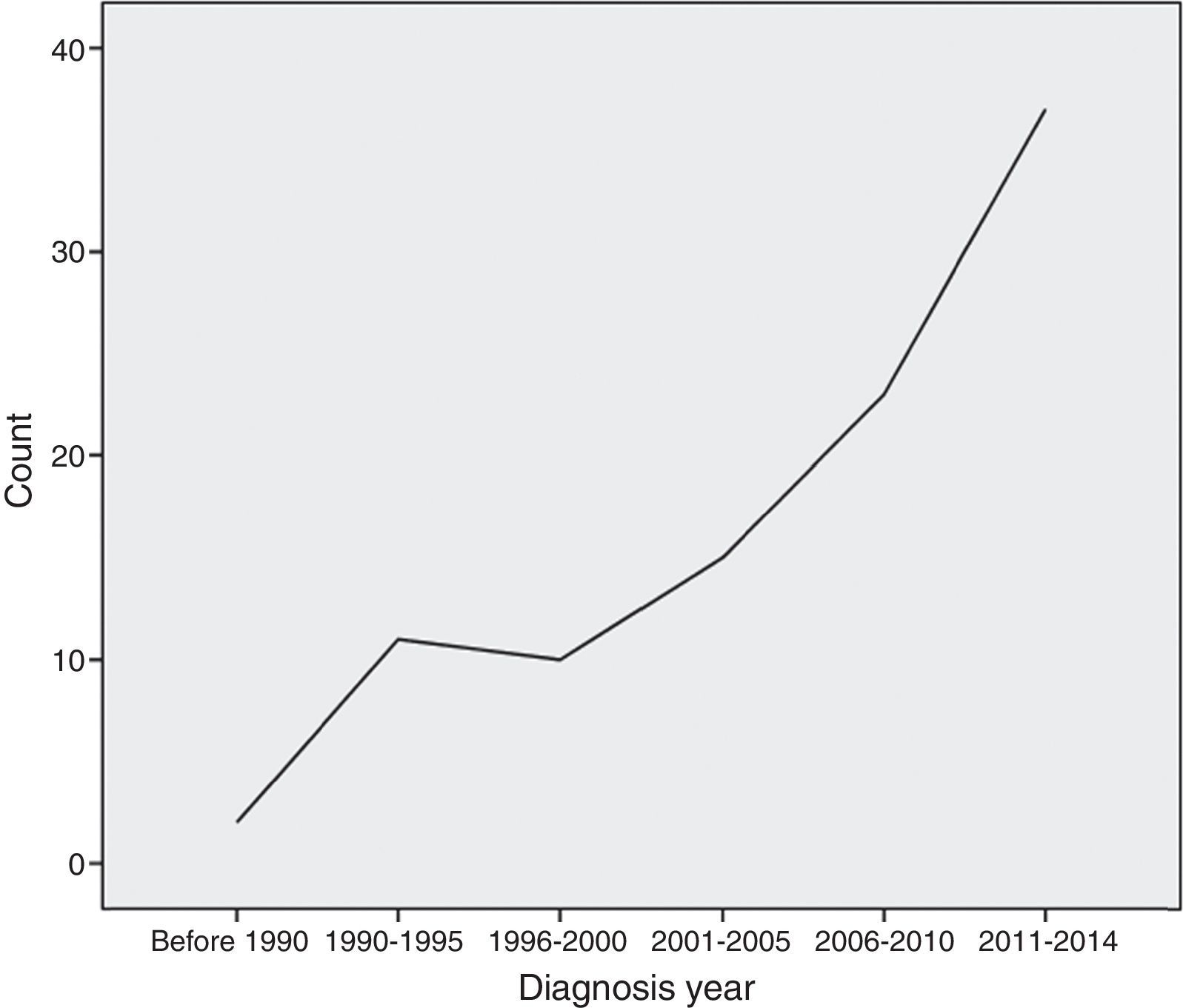
MethodThis was a single-centre retrospective study carried out at Allergy and Clinical Immunology Department of Rasool-E-Akram, an 830-bed tertiary referral hospital, in Tehran, the capital city of Iran, from January 1989 to July 2014 (Fig. 1).
, Tehran, Iran between 1990 and 2014.")
The diagnosis of PID was made after secondary forms of immunodeficiency were ruled out and according to clinical and laboratory findings and diagnostic criteria of IUIS, PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies).1,7,8 Genetic testing (to confirm the diagnosis of X linked agammaglobulinaemia, X linked hyper IgM, LRBA deficiency, DOCK8 deficiency, STAT1 and HAX1) was done as part of genetic consultation with Research Center for Immunodeficiency in Children's Medical Center in Tehran University of Medical Sciences. Only patients with a definite diagnosis of PID were enrolled for the purpose of the study. Demographic, clinical and immunological data were collected using a standardised questionnaire.
Serological and immunological laboratory testing included quantitative serum immunoglobulin levels, IgG subclass levels, pre- and post-immunisation antibody titres to Pneumococcus, Diphtheria and Tetanus, isohaemagglutinin titres, flow cytometric assessment of lymphocyte subsets, lymphocyte transformation test, nitroblue tetrazolium test, complement component (C3, C4, CH50) tests and delayed type hypersensitivity skin testing to Candida antigen.
The majority of the patients were diagnosed at Rasool-E-Akram Hospital, whereas some of the older patients were initially diagnosed at other institutes (Mofid Hospital, Ali-Asghar Hospital and Children's Medical Center) and were referred for continued treatment and follow-up. The patients were routinely visited at 1–3 months interval.
Statistical analysesStatistical analyses were performed using STATA 10 software. Quantitative data were reported as mean±SD and descriptive variables as proportion. T-test was used to compare means and chi-square test was used to compare proportions. P-values less than 0.05 were considered statistically significant.
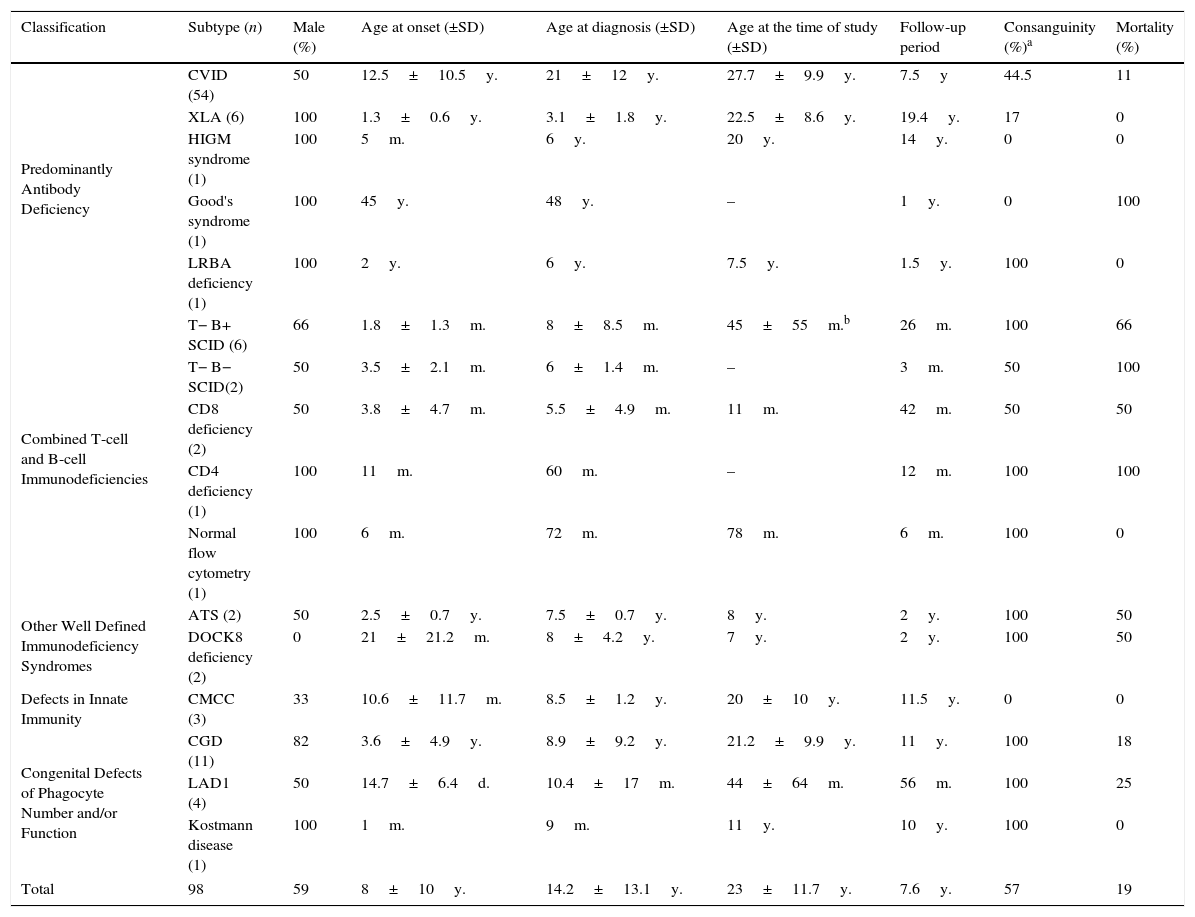
ResultsNinety-eight patients were diagnosed with 15 different disorders in five main groups. Table 1 describes demographic characteristics and mortality rates of these patients. Nineteen patients (19%) aged 17.5±18.2 years died during the follow-up period due to severe infections, malignancies, graft versus host disease and hepatic failure.
Demographic characteristics and outcomes of ninety eight Iranian patients with primary immune deficiency.
| Classification | Subtype (n) | Male (%) | Age at onset (±SD) | Age at diagnosis (±SD) | Age at the time of study (±SD) | Follow-up period | Consanguinity (%)a | Mortality (%) |
|---|---|---|---|---|---|---|---|---|
| Predominantly Antibody Deficiency | CVID (54) | 50 | 12.5±10.5y. | 21±12y. | 27.7±9.9y. | 7.5y | 44.5 | 11 |
| XLA (6) | 100 | 1.3±0.6y. | 3.1±1.8y. | 22.5±8.6y. | 19.4y. | 17 | 0 | |
| HIGM syndrome (1) | 100 | 5m. | 6y. | 20y. | 14y. | 0 | 0 | |
| Good's syndrome (1) | 100 | 45y. | 48y. | – | 1y. | 0 | 100 | |
| LRBA deficiency (1) | 100 | 2y. | 6y. | 7.5y. | 1.5y. | 100 | 0 | |
| Combined T-cell and B-cell Immunodeficiencies | T− B+ SCID (6) | 66 | 1.8±1.3m. | 8±8.5m. | 45±55m.b | 26m. | 100 | 66 |
| T− B− SCID(2) | 50 | 3.5±2.1m. | 6±1.4m. | – | 3m. | 50 | 100 | |
| CD8 deficiency (2) | 50 | 3.8±4.7m. | 5.5±4.9m. | 11m. | 42m. | 50 | 50 | |
| CD4 deficiency (1) | 100 | 11m. | 60m. | – | 12m. | 100 | 100 | |
| Normal flow cytometry (1) | 100 | 6m. | 72m. | 78m. | 6m. | 100 | 0 | |
| Other Well Defined Immunodeficiency Syndromes | ATS (2) | 50 | 2.5±0.7y. | 7.5±0.7y. | 8y. | 2y. | 100 | 50 |
| DOCK8 deficiency (2) | 0 | 21±21.2m. | 8±4.2y. | 7y. | 2y. | 100 | 50 | |
| Defects in Innate Immunity | CMCC (3) | 33 | 10.6±11.7m. | 8.5±1.2y. | 20±10y. | 11.5y. | 0 | 0 |
| Congenital Defects of Phagocyte Number and/or Function | CGD (11) | 82 | 3.6±4.9y. | 8.9±9.2y. | 21.2±9.9y. | 11y. | 100 | 18 |
| LAD1 (4) | 50 | 14.7±6.4d. | 10.4±17m. | 44±64m. | 56m. | 100 | 25 | |
| Kostmann disease (1) | 100 | 1m. | 9m. | 11y. | 10y. | 100 | 0 | |
| Total | 98 | 59 | 8±10y. | 14.2±13.1y. | 23±11.7y. | 7.6y. | 57 | 19 |
Note. CVID: Common Variable Immune Deficiency; XLA: X Linked Agammaglobulinaemia; HIGM: Hyper-IgM; LRBA: LPS responsive beige-like anchor; SCID: severe combined immunodeficiency; ATS: Ataxia-Telangiectasia syndrome; DOCK8: Dedicator of Cytokinesis 8; CMCC: Chronic Mucocutaneous Candidiasis; CGD: chronic granulomatous disorder; LAD: leucocyte adhesion molecule deficiency; y.: years; m.: months; d.: days.
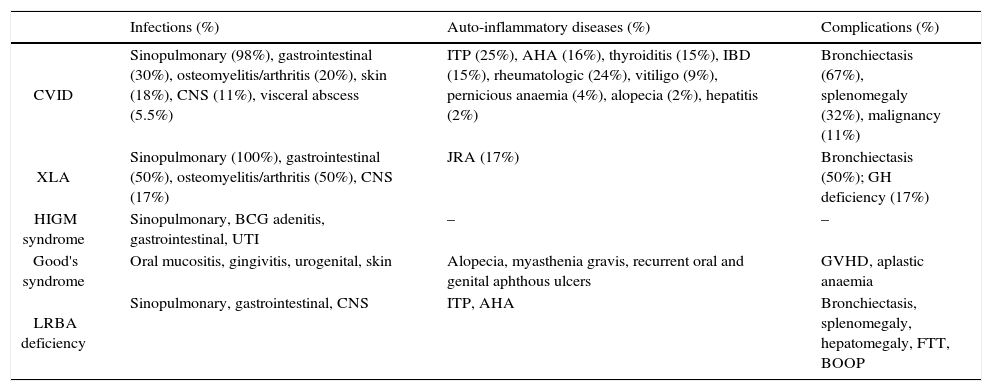
PAD was the most common diagnosis and included: common variable immunodeficiency (CVID), X-linked agammaglobulinaemia (XLA), hyper-IgM (HIGM) syndrome, thymoma with hypogammaglobulinaemia (Good's syndrome), and hypogammaglobulinaemia with mutations in lipopolysaccharide-responsive beige-like anchor (LRBA) gene (Tables 1 and 2). All patients were treated with intravenous immune globulin (IVIG) at least on a monthly basis, and those with severe bronchiectasis or recurrent bacterial infections were treated with prophylactic antibiotics.
Clinical manifestations and complications of 63 Iranian patients with predominantly antibody deficiency.
| Infections (%) | Auto-inflammatory diseases (%) | Complications (%) | |
|---|---|---|---|
| CVID | Sinopulmonary (98%), gastrointestinal (30%), osteomyelitis/arthritis (20%), skin (18%), CNS (11%), visceral abscess (5.5%) | ITP (25%), AHA (16%), thyroiditis (15%), IBD (15%), rheumatologic (24%), vitiligo (9%), pernicious anaemia (4%), alopecia (2%), hepatitis (2%) | Bronchiectasis (67%), splenomegaly (32%), malignancy (11%) |
| XLA | Sinopulmonary (100%), gastrointestinal (50%), osteomyelitis/arthritis (50%), CNS (17%) | JRA (17%) | Bronchiectasis (50%); GH deficiency (17%) |
| HIGM syndrome | Sinopulmonary, BCG adenitis, gastrointestinal, UTI | – | – |
| Good's syndrome | Oral mucositis, gingivitis, urogenital, skin | Alopecia, myasthenia gravis, recurrent oral and genital aphthous ulcers | GVHD, aplastic anaemia |
| LRBA deficiency | Sinopulmonary, gastrointestinal, CNS | ITP, AHA | Bronchiectasis, splenomegaly, hepatomegaly, FTT, BOOP |
Note. CVID: Common Variable Immune Deficiency; XLA: X Linked Agammaglobulinaemia; HIGM: Hyper-IgM; LRBA: LPS responsive beige-like anchor; CNS: central nervous system; UTI: urinary tract infection; ITP: idiopathic thrombocytopenic purpura; AHA: autoimmune haemolytic anaemia; IBD: inflammatory bowel disease; JRA: juvenile rheumatoid arthritis; GH: growth hormone; GVHD: graft versus host disease; FTT: failure to thrive; BOOP: bronchiolitis obliterans organising pneumonia.
CVID – Recurrent infections (85%) and autoimmune manifestations (15%) were first presentations in these patients. The age at onset in patients with parental consanguinity was significantly lower than the others (7.7±7.8 versus 16.3±11 years; P value=0.001). There was no relationship between CVID phenotypes or complications such as autoimmunity or malignancy and parental consanguinity. A total of 12 (22%) patients had severe viral infections, including cytomegalovirus (gastroenteritis, colitis, pneumonia), varicella zoster virus (disseminated varicella, pneumonia), human papillomavirus (recurrent warts), herpes simplex virus (severe skin infections, gingivostomatitis), and John Cunningham virus (progressive multifocal leukoencephalopathy). Fungal infections occurred in three patients and included recurrent oral thrush and Pneumocystis jiroveci pneumonia. Twenty-five (45%) patients developed autoimmune manifestations, among them 12 patients had multiple autoimmune diseases. Six patients developed seven malignancies, including Hodgkin's disease (in three cases), pre B cell leukaemia, breast cancer, gastrointestinal adenocarcinoma and brain cancer. Six patients aged 35.4±17.2 years died during the follow-up period of 4.5±4.3 years due to sepsis (n=2), malignancy (n=2), viral CNS infection (n=1), and hepatic failure (n=1).
XLA – Six males aged 22.5±8.6 years were followed for mean 19.4±6.9 years. The diagnosis was confirmed with mutational analysis of the BTK gene in all the cases. One patient was diagnosed and treated for isolated growth hormone deficiency. No patients died during the follow-up period.
HIGM syndrome – This was diagnosed in one male patient with a history of recurrent infections and hypogammaglobulinaemia and the diagnosis was confirmed by mutational analysis of the CD40 ligand gene (CD40L – Exon 5 – c.187C>T). At the time of study, the patient was 20 years old with no history of severe infections or complications after proper therapeutic interventions were initiated.
Good's syndrome – One male patient with a history of myasthenia gravis and thymoma, undergoing thymectomy, was diagnosed with Good's syndrome following the development of clinical and laboratory features of hypogammaglobulinaemia (Table 2). The patient died aged 49 years of severe acute graft-versus-host disease syndrome following transfusion of irradiated blood products.
LRBA deficiency – One seven-year-old male patient, referred at six years of age with a history of recurrent infections, agammaglobulinaemia and absent B-lymphocyte, who was diagnosed with genetic analysis. He developed severe pneumonia with Pneumocystis jiroveci and CMV six months after diagnosis, and was later diagnosed with bronchiolitis obliterans with organising pneumonia.
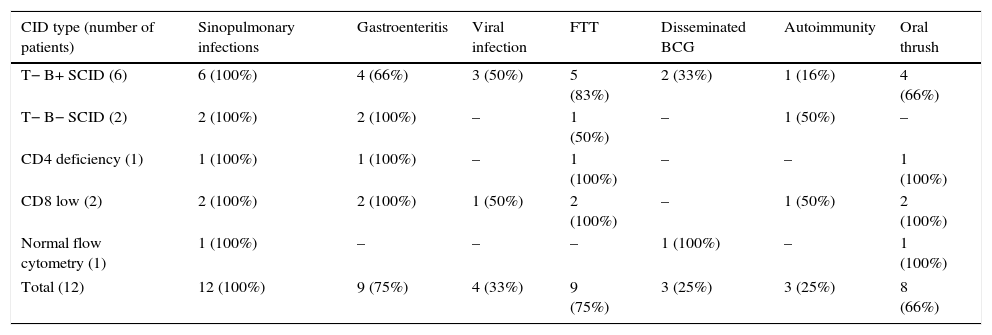
Combined T-cell and B-cell immunodeficienciesTwelve patients were diagnosed with CID and of them eight had severe combined immunodeficiency (six T−B+ and two T−B−) and four had less severe CID (Table 1). The most common presentations were pneumonia (100%), gastroenteritis (75%), chronic oral thrush (66%), and disseminated BCG infection after vaccination (25%). Severe viral (CMV, HSV) and fungal (Pneumocystis jiroveci) infections were diagnosed in 33 and 25% of patients respectively (Table 3). Three patients developed autoimmune disorders, including ITP, vasculitis and eczema. Lymphopenia was observed in five (42%) patients: two (33%) of T−B+, two (100%) of T−B− and one patient (100%) with CD4 deficiency. A positive family history of diagnosed or suspected immune deficiency was reported in three patients. Patients were treated with IVIG, prophylactic antibiotics, antifungals and isoniazid (because of BCG vaccination) and were considered for haematopoietic stem-cell transplantation (HSCT). Only one patient (T−B+) had HSCT from a matched sibling donor after low intensity conditioning regimen with fludarabine, melphalan and anti thymocyte globulin therapy and was seven years old at the time of study with no long-term complication following bone marrow transplant. Eight patients died at the age of 36±34 months due to severe pneumonia, gastroenteritis, sepsis and disseminated BCG infection. Age at expiration was 29±31 months in T−B+, 8.5±3.5 months in T−B−, 72 and 84 months in CD4 deficiency and CD8 low patients respectively.
Common clinical manifestations in twelve Iranian combined immunodeficiency disorders.
| CID type (number of patients) | Sinopulmonary infections | Gastroenteritis | Viral infection | FTT | Disseminated BCG | Autoimmunity | Oral thrush |
|---|---|---|---|---|---|---|---|
| T− B+ SCID (6) | 6 (100%) | 4 (66%) | 3 (50%) | 5 (83%) | 2 (33%) | 1 (16%) | 4 (66%) |
| T− B− SCID (2) | 2 (100%) | 2 (100%) | – | 1 (50%) | – | 1 (50%) | – |
| CD4 deficiency (1) | 1 (100%) | 1 (100%) | – | 1 (100%) | – | – | 1 (100%) |
| CD8 low (2) | 2 (100%) | 2 (100%) | 1 (50%) | 2 (100%) | – | 1 (50%) | 2 (100%) |
| Normal flow cytometry (1) | 1 (100%) | – | – | – | 1 (100%) | – | 1 (100%) |
| Total (12) | 12 (100%) | 9 (75%) | 4 (33%) | 9 (75%) | 3 (25%) | 3 (25%) | 8 (66%) |
CID: combined immunodeficiency disorders; SCID: severe combined immunodeficiency disorders; FTT: failure to thrive; BCG: bacillus calmette-guerin.
Ataxia Telangiectasia Syndrome (ATS) – Two patients were diagnosed with neurologic symptoms, telangiectasia and history of recurrent pneumonia and gastroenteritis. Both were treated with IVIG infusion, prophylactic antibiotics and pulmonary care. One of the patients with history of UTI, sepsis, severe bronchiectasis, pulmonary hypertension and respiratory failure, died at 10 years of age after a follow-up period of three years. The second patient, an eight-year-old boy at the time of study, was diagnosed with large B cell lymphoma and was under chemotherapy.
Dedicator of Cytokinesis 8 (DOCK8) deficiency – One female patient was referred with history of severe eczema, recurrent urticaria and angioedema, recurrent sinopulmonary infections and BCG adenitis. Laboratory evidences included eosinophilia and normal immunoglobulin levels on multiple, separate occasions, except for an elevated IgE level of 534IU/mL (reference range: <81) reported on one occasion. The diagnosis was confirmed by genetic analysis that showed large homozygous deletion in exon 6 through 15 of the DOCK8 gene. She was treated symptomatically and was candidate for HSCT. Her sister had a similar history and died at 11 years of age due to undiagnosed brain tumour.
Chronic Mucocutaneous CandidiasisAutoimmune manifestations of three CMCC patients included: hypothyroidism, hypoparathyroidism, Addison disease, autoimmune hepatitis, ITP, autoimmune haemolytic anaemia, alopecia and vitiligo. Increased susceptibility to bacterial infections, including sinopulmonary infections, perianal abscess, urinary tract infection, gingivitis and diarrhoea were identified in these patients during the follow up. One patient with heterozygous gain-of-function mutation in STAT1, who was 30 years old at the time of study, had developed bronchiectasis and pulmonary hypertension due to recurrent severe pulmonary infections. All patients received antifungal prophylaxis.
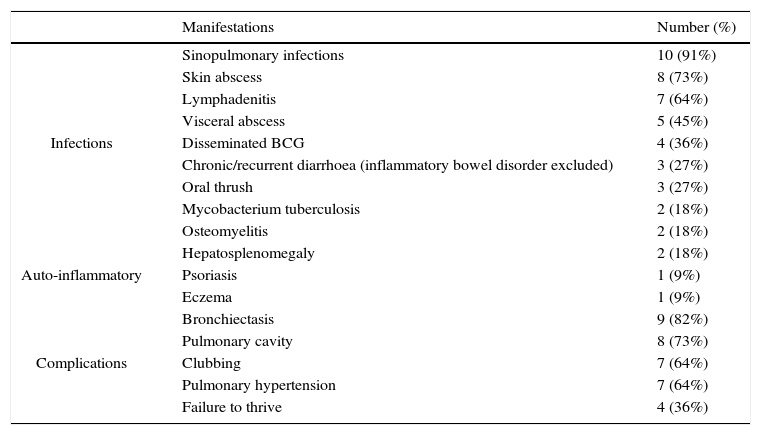
Chronic granulomatous disorder (CGD) – The clinical manifestations and complications of eleven patients with CGD are summarised in Table 4. The inheritance pattern in four patients was X linked and in the others was autosomal recessive, according to pedigrees and maternal NBT results. Six patients had a positive family history. Patients were treated with itraconazole and co-trimoxazole prophylactically; however, not all patients were compliant to this regimen due to its cost and lack of insurance. Inaccessibility and cost precluded the use of interferon-gamma (IFN-γ) in any of the patients. None of the patients received bone marrow transplant. Two patients died of severe pneumonia and respiratory failure during the follow-up period.
Clinical manifestations and complications of eleven Iranian patients with chronic granulomatous disease.
| Manifestations | Number (%) | |
|---|---|---|
| Infections | Sinopulmonary infections | 10 (91%) |
| Skin abscess | 8 (73%) | |
| Lymphadenitis | 7 (64%) | |
| Visceral abscess | 5 (45%) | |
| Disseminated BCG | 4 (36%) | |
| Chronic/recurrent diarrhoea (inflammatory bowel disorder excluded) | 3 (27%) | |
| Oral thrush | 3 (27%) | |
| Mycobacterium tuberculosis | 2 (18%) | |
| Osteomyelitis | 2 (18%) | |
| Auto-inflammatory | Hepatosplenomegaly | 2 (18%) |
| Psoriasis | 1 (9%) | |
| Eczema | 1 (9%) | |
| Complications | Bronchiectasis | 9 (82%) |
| Pulmonary cavity | 8 (73%) | |
| Clubbing | 7 (64%) | |
| Pulmonary hypertension | 7 (64%) | |
| Failure to thrive | 4 (36%) |
Leucocyte adhesion molecule deficiency type 1 (LAD1) – All patients were presented with omphalitis and delayed separation of umbilical cord. Other manifestations included cellulitis, peritonitis, chronic diarrhoea, failure to thrive, septic arthritis, recurrent gingivitis, conjunctivitis and sepsis. Two cases were siblings with CD18 of 2%; one died of extra-nodal large B cell lymphoma at 11 years of age and the other one was 10 years old at the time of study. The other two patients had undetectable CD18 levels and were 4 and 12 months old, respectively. The patients were under prophylactic antibiotics while waiting to receive HSCT.
Kostmann disease – One male patient with a history of neonatal sepsis, recurrent pneumonia, otitis media, cellulitis, skin abscess and gingivitis was diagnosed with congenital neutropenia due to mutation in HAX1 gene. The patient treated with prophylactic antibiotics and G-CSF, with only mild neutropenia and no serious complication.
DiscussionSince the first report of Iranian PID registration in 2002, more than 1600 PID cases were reported from 14 Iranian academic medical centres.1 An increasing number of experts in the field of clinical immunology, as well as increasing knowledge and awareness of physicians, has enhanced early diagnosis and management of this rare but significant group of diseases.4,9
Consanguinity – In this report, the total parental consanguinity rate was 57%. All patients diagnosed with phagocytic disorders and well defined syndromes had a positive history of parental consanguinity. This rate was 83% and 41% among patients diagnosed with CID and PAD, respectively. Consanguinity increases the risk of autosomal recessive diseases, including PID; the closer the relationship, the higher the risk.10 In regions where consanguineous marriage is intrinsic to the culture, the frequency and type of genetic defects causing PID differ in comparison with those in other populations.11–14
Clinical phenotypes and the severity of CVID have been shown to be related to parental consanguinity in some registries,15,16 but in our CVID cases, there was no relationship between CVID phenotypes or complications such as autoimmunity or malignancy and parental consanguinity.
Age at onset – Most types of PID begin early in life. In our cohort, patients with CID, LAD1 and Kostmann disease were symptomatic in the first months of life, whereas, patients with PAD (except for CVID), ATS, DOCK8 deficiency, CMCC, and CGD were symptomatic in the first few years of life. This was similar to previous reports from Iran and other countries.1,4,17–22 In patients with CVID, the mean age at onset was 12.5 years, which falls in line with that of western countries in the second or third decades of life.5 However, it should be underscored that the mean age at onset of patients with CVID was lower than three years in reports from other centres in Iran. This could be due to the high rate of parental consanguinity and complex inherited genetic basis in Iranian CVID patients, as we reported significantly lower age at onset in patients with a positive history of parental consanguinity than the others (7.7 versus 16.3 years). Moreover, our centre is located at a tertiary general hospital, unlike other centres in Iran that are mainly affiliated to children's hospitals, which could explain the higher age at onset of referral patients at our institution. We previously described that higher age of onset of CVID is significantly associated with the higher risk of malignancy and autoimmune disorders.14
Age at diagnosis and diagnostic delay – In our cohort of patients, the mean diagnostic delay was 6.1±7 years (1 month to 31 years), and those with less severe forms of PID, such as PAD and CMCC, had more prolonged diagnostic delay. Patients with CVID had a mean diagnostic delay of 8.5±7.9 years, while it was only 2±1.4 years in other PAD subtypes. The majority of patients with CID had a diagnostic delay of only a few months, except for two patients with CD4 deficiency and normal flow cytometry. Most patients with LAD1 and Kostmann disease were diagnosed in infancy. Patients with well-defined immunodeficiency syndromes were diagnosed in childhood with 4±2 years of diagnostic delay. The delayed diagnosis and treatment of PID is associated with an increased overall risk of disease-related complications.1,4,23
Infections – In patients with PAD, sinopulmonary infections were the most common bacterial infections. Viral infections were diagnosed in 22% of CVID patients and the patient with LRBA deficiency. Fungal infections occurred in three of the CVID patients, as well as patients with LRBA deficiency and Good's syndrome. These infections can be due to associated abnormalities in T cell and innate immunity.24 None of the XLA patients had history of severe viral or fungal infection.
Patients with CID are prone to bacterial, fungal and viral infections.19,25,26 The most common infections in patients with CID were pneumonia, gastroenteritis, oral thrush. Severe viral and fungal infections were reported in 25% of patients. BCG vaccine is given once at birth in most developing countries, including Iran and disseminated BCG infection is a common feature in immune deficiency syndromes, including CID, CGD and defects in the IL-12/IFN-γ axis.27 In our study, 25% of CID and 36% of CGD patients had disseminated BCG infection.
Consistent with other reports, the most common infectious presentation in our cohort of CGD patients was sinopulmonary infections, followed by lymphadenitis, and skin and visceral abscess (Table 4). Mycobacterium tuberculosis (pulmonary infection, lymphadenitis) was diagnosed in two patients. Mycobacterium tuberculosis can be the first manifestation of CGD in endemic regions.28 All four patients with LAD1 initially presented with omphalitis and delayed umbilical separation in the first weeks of life. Other infections included cellulitis, peritonitis, chronic diarrhoea and septic arthritis, and were similar to previous reports.18 Severe congenital neutropenia is clinically characterised by omphalitis, pneumonia and gingivitis caused by resident bacteria of the skin, mouth, and oropharynx.21 Similarly, the patient reported herein presented with neonatal sepsis, respiratory tract and mucocutaneous infections.
CMCC is characterised with chronic non-invasive Candida infections of the skin, nails, and mucous membranes, in association with a variety of clinical features, including increased susceptibility to bacterial and viral infections recommended.26 We reported recurrent sinopulmonary infections, perianal abscess, urinary tract infection, and gingivitis in patients diagnosed with CMCC.
In patients with ATS, immune deficiency is quite variable but often manifests as recurrent sinopulmonary infections. Infections outside of the respiratory tract are generally not increased in frequency, and opportunistic infections rarely occur. Pulmonary infections may be the result of underlying immunodeficiency and aspiration.22,29 Similarly, both patients with ATS had history of recurrent pneumonia and gastroenteritis. Patients with DOCK8 deficiency develop recurrent upper and lower respiratory tract infections, extensive cutaneous viral and bacterial infections, mucosal or nail candidiasis and otitis externa.30 The reported DOCK8 deficient patients reported herein had recurrent sinopulmonary infections and BCG adenitis, with no history of severe viral infections.
Autoimmunity – Autoimmunity and abnormal inflammation has often been observed in association with different forms of PID.3 Autoimmune disorders may be the first manifestation of the disease in CVID patients.5,6 In our study 25 (46%) CVID patients had autoimmune disorders and ITP was the most common diagnosis. Autoimmune disorders were the first signs of immunodeficiency in eight (15%) patients. Twelve (22%) patients had multiple autoimmune disorders. Patients with Good's syndrome and LRBA deficiency were diagnosed with multiple autoimmune disorders, and one out of six patients with XLA had history of juvenile rheumatoid arthritis. Three (25%) patients with CID developed ITP, vasculitis and eczema. Severe, chronic eczema was the main autoimmune feature of the two DOCK8 deficient patients. Patients with CMCC developed multiple autoimmune disorders including endocrinopathies, autoimmune hepatitis, ITP, AHA, alopecia and vitiligo. Two (18%) patients with CGD were diagnosed with psoriasis and eczema.
Treatment protocols – Lack of or inadequate medical insurance coverage for the treatment of PID manifestations and complications in Iran has limited the extent of therapeutic interventions to symptomatic and supportive treatments.1 The cohort of 63 PAD patients in this study was treated with IVIG replacement at least monthly, in addition to treatment of any active infections and/or autoimmune disorders as was recommended.5,26,31 The goal of therapy was to maintain IgG trough levels (drawn just before the next infusion) of at least 500mg/dL up to the end of 2012, which was later increased to 600–700mg/dL.
The treatment of patients with CID includes HSCT and gene therapy and earlier detection of the disease and treatment in few months after birth significantly improves the outcomes.25,26,31 In this study, only one patient (T−B+) underwent a bone marrow transplant, who showed no evidence of long-term complications at the time of study. The remaining three patients were treated with antimicrobials and IVIG while waiting to receive HSCT.
The two patients with ATS reported herein were treated with IVIG replacement therapy and antibiotics as recommended.22,26,29,31 Treatment of DOCK8 deficient patients are mainly supportive, and HSCT remains the only definitive option.30 Antibiotic prophylaxis, skin care and prompt treatment of skin infections were supportive treatment in these patients and the living case is waiting to receive HSCT.
Management of patients with CMCC included antifungal therapy and treatment of associated endocrinopathies and autoimmune abnormalities, as well as antibiotic therapy as recommended.26
The cornerstones of management of CGD are prophylaxis with antibacterial, antifungal and IFN-γ and attempts to early diagnosis and aggressive treatment of infectious complications. However, successful HSCT can be a definitive cure for CGD.26,31,32 In our study, costs of antifungal prophylaxis resulted in poor patient compliance. No patients received IFN-γ and HSCT during the follow-up period because of unavailability. Mild to moderate LAD1 usually responds to antibiotic therapy, while HSCT is recommended in severe cases (CD11a CD18<1%).26,31 All four patients with LAD1 were treated for acute infections and received prophylactic antibiotics. The two patients with the severe form of the disease were potential candidates for HSCT. The patient with Kostmann disease was treated with G-CSF, as well as antibiotics for prophylaxis and treated for acute infections. In severe cases of congenital neutropenia, who are unresponsive to G-CSF or complicated with lymphoproliferative disorders, HSCT is recommended.21,26,31
Complications and mortality – Bronchiectasis was the most common complication among patients with PAD (Table 2). Other common complications in patients with CVID were splenomegaly (32%) and malignancy (11%). Malignancies are reported in 2.5–8% of CVID patients, with non-Hodgkin's lymphoma being the most common reported malignancy.5,33 During the mean follow-up period of 8.5 years, 11% of CVID patients died, compared to a mortality rate of 35% during a mean follow-up period of 6.5 years in an Iranian study performed in 2007.34 The decrease in the mortality rate could be due to earlier diagnosis and improved availability of IVIG and supportive care. Similar to previous reports,35 patients with XLA had lower morbidity and mortality compared to patients with CVID: six patients with XLA were followed up for a mean period of 19.4 years with no severe complications. One of the patients had XLA and growth hormone deficiency. This was first described by Fleisher et al. in 1980 and since that time other patients have been reported with this syndrome36,37 and clinicians should follow the stature growth in their XLA patients. The patient with Good's syndrome died of acute graft-versus-host disease syndrome following the transfusion of irradiated blood products for symptomatic treatment of aplastic anaemia. This emphasizes the importance of awareness of physicians about the risk of GVHD in cellular immunodeficiency conditions such as Good's syndrome.38 We reported a case of LRBA deficiency, a rare autosomal recessive CVID-like syndrome,39 which was complicated with bronchiolitis obliterans with organising pneumonia.
The only patient reported with HIGM syndrome herein, was followed for 14 years and was 20 years old at the time of study, with no history of severe infection or disease related complication.
Patients diagnosed with CID often die in infancy or the first few years of life, despite supportive therapy, unless treated with HSCT from an appropriate donor.25,26 In our cohort of CID patients, except for one who underwent HSCT, only three patients with milder phenotype survived for years.
The major complications of patients with ATS are progressive neurologic deterioration, respiratory syndrome, chronic lung diseases and malignancies,22,29 which rarely allow survival beyond the second or third decade of life. In our study, one patient died at 10 years of age due to poor family support and severe lung disease. The second patient was diagnosed with and treated for lymphoma at the time of study. DOCK8 deficiency has a relatively poor prognosis due to life threatening infections, malignancy or cerebral complications such as CNS vasculitis or stroke.30 One of the patients with DOCK8 deficiency died of an undiagnosed brain mass at 11 years of age.
One patient with heterozygous gain-of-function mutation in STAT1 developed chronic lung disease and pulmonary hypertension because of recurrent pneumonia. Severe phenotypes in these patients were reported previously.40
In our cohort of CGD patients, the majority developed chronic lung disease with severe bronchiectasis, cavity formation, pulmonary hypertension and clubbing. These morbidities were previously reported in patients with CGD32; however, unaffordable medicine prices and poor availability made these severe complications much more common among our patients. Without successful HSCT, infections are the major cause of death in patients with severe LAD1.18,26 In our study, one of the patients with moderate LAD1 died of lymphoma at 11 years of age. Two infants with undetectable CD18 levels are waiting for HSCT. The only patient with Kostmann disease was 12 years old and in good condition at the time of study with no severe complication. Patients with a good response to G-CSF have acceptable prognosis; however, they should be observed for infections and lymphoproliferative disorders.21
ConclusionsPhysicians’ awareness of PID has been rising dramatically in certain regions of the world, ensuring an increasing number of patients being diagnosed and treated. However, a large number of patients do not have access to effective treatment services and suffer from lack of health insurance and weak or absent social supports.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in this study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe authors have no conflict of interest to declare.
