


INTRODUCCION
La primera descripción del síndrome de Hiper IgE data del año 1966, cuando Davis et al1 relatan un grupo de mujeres jóvenes, que padecen eccema y abscesos fríos recurrentes en piel, tejido celular subcutáneo y nódulos linfáticos, de etiología estafilococica. A esta entidad primera se le denominó síndrome de Job.
La asociación de infecciones recurrentes y niveles elevados de Ig E fueron descritos en el año 1972 por Buckley et al2 en dos adolescentes que presentaban dermatitis crónica, infecciones bacterianas recurrentes en piel y pulmón con facies tosca3. En 1974 este síndrome se incluye dentro del grupo de las inmunodeficiencias primarias. Revisiones posteriores, como la de Leung y Geha de 1988, se centran en la descripción de las principales características clínicas y alteraciones inmunitarias4. Aunque en la mayoría de los casos no existen antecedentes familiares de infecciones recurrentes ni de síndrome de Hiper Ig E, en estudios recientes se ha descrito un defecto genético en el brazo largo del cromosoma 4 en algunas familias afectas.En cualquier caso, la expresión de la entidad sugiere una herencia autosómica dominante con penetrancia variable5. Este síndrome continúa siendo controvertido y se define por la existencia de criterios clínicos compatibles y niveles elevados de IgE por encima de 2.000 UI/ml6.
CASO CLINICO
Niña de 5 años de edad remitida por su pediatra por presentar niveles elevados y crecientes de IgE sérica desde los 3 años de edad (7.419 U/ml). En el momento de la consulta la paciente está febril, con otorrea bilateral, linfadenopatías cervicales y celulitis de la rodilla izquierda.
Antecedentes familiares no valorables. Primera hija de matrimonio no consanguíneo. Parto a término, RN de peso adecuado para edad gestacional. Retraso en la caída del cordón umbilical (36 días de vida). Desde los 6 meses de edad: otitis supuradas con frecuencia mensual y buena respuesta a la antibióticoterapia, pero que persisten a pesar de efectuar adenoidectomía y colocación de drenajes transtimpánicos a los 3 años de edad.
Desde el año de vida: dermatitis flexural y de cuero cabelludo y foliculitis ocasional (1-2 por año) que se resuelven con tratamiento antibiótico tópico. Vulvovaginitis de repetición. Varicela a los dos años y seis meses sin complicaciones. Ingreso por una gastroenteritis por Salmonella a los 3 años, de evolución sin complicaciones. Después de los 4 años de edad ha tenido uno o dos episodios de bronquitis que evolucionaron favorablemente con antibióticos. Herpes zoster a los 5 años.
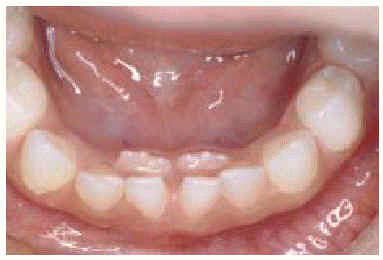
Exploración física: desarrollo pondoestatural normal. Buen estado general, febril 38 °C. Xerosis, lesiones papulares de pequeño tamaño de características eccematosas con excoriación en cuello, axila y periné con afectación interdigital de dedos del pie. Onicomicosis en primeros dedos de ambos pies. Onfalitis exudativa. ORL: otorrea serosanguinolenta bilateral, linfadenopatías latero-cervicales altas aisladas, mal delimitadas, no dolorosas y de gran tamaño (2 x 3 cm en región lateral izquierda y 4 x 5 cm en región lateral derecha). Orofaringe congestiva, amígdalas sin exudados, lisas y brillantes. Persistencia de la dentición temporal de incisivos inferiores. Presencia de dentición permanente (doble arcada dentaria) (fig. 1). Genitourinario: vulvovaginitis. Tumefacción, eritema y aumento de calor de rodilla izquierda sin limitaciones de la movilidad articular.
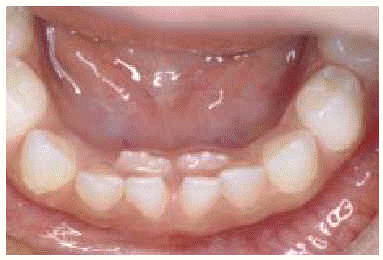
Figura 1.--Doble arcada dental.
MATERIAL Y MÉTODOS
Hemograma: anemia normocítica, normocrómica (hematíes: 3,63 mill/ml, Hb 10,5 g/dl, HTC 31,5 %, VCM, HMC, normales). Leucocitos 12.600/ml (8 %L, 10 %M, 69 %N, 7 %C, 6 %E, 0 %B). VSG 90mm, PCR 164,4 mg/L. ASLOS: < 200 U/mL.
Inmunidad humoral: IgG: 23.000 mg/L ↑, IgA: 4.220 mg/L ↑, IgM: 878 mg/L, IgE total: 23.969 kUI/L ↑, IgD 440 U/ml ↑ . Subclases de IgG: IgG1:9.280 mg/L ↑, IgG2: 10.800 mg/L ↑, IgG3: 89 mg/L, IgG4: 1.860mg/L ↑. Prick test a neumoalergenos y alimentos: negativo. Isohemaglutininas: anti-B positivo. Anticuerpos frente a: rubéola, virus varicela zoster, antitetánicos, antipoliomielitis antidiftérica, neumocócicos, y H. infuenzae positivos. Anti- Candida albicans negativo.
Inmunidad celular: Poblaciones linfoides: Linfocitos: B: 15 %, T3 43 %, T4 63 %, T8 34 %, T4/T8:1,85. TTL frente a mitógenos inespecíficos (ConA, PHA, PWM): normal. Pruebas in vivo de inmunidad celular (Multitest): candidina: negativas.
Inmunidad inespecífica: Presencia de moléculas LAD I y II, Complemento: C'3 1.080 mg/L, C'4 243 mg/L, CH 50: 40U/ml, Test del NBT: 10 %3-10.
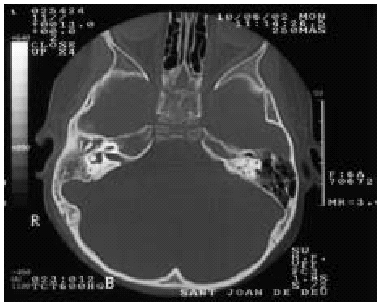
Otras exploraciones: Ac anti VIH: negativo. Exploraciones radiológicas: TAC mastoides: ocupación de caja timpánica, antro y mastoides derecha, con aumento de densidad de partes blandas, sin erosiones óseas, compatible con otomastoditis derecha (fig. 2).
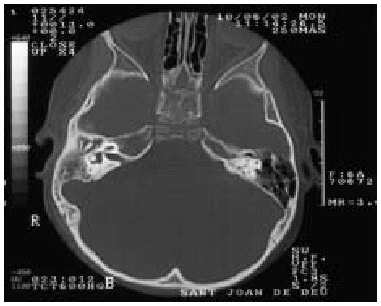
Figura 2.--Ocupación de caja timpánica, antro y mastoides derecha, con aumento de la densidad de partes blandas, sin erosiones óseas, compatible con otomastoiditis derecha.
Engrosamiento de membrana timpánica izquierda. TAC de mastoides de control, normal. Densitometría ósea: normal. Microbiología: cultivo de secreción ótica (recogida por paracentesis): Candida albicans; de exudado vaginal-perineal: Enterococcus faecium; exudado umbilical: Enterococcus faecium.
Ante el diagnóstico de mastoiditis por Candida albicans, la paciente se trata con anfotericina B liposomal, con la que comienza a disminuir la secreción ótica. Después de conocer los resultados de los cultivos de exudado de ombligo y región vaginal, en los que se aíslan colonias de enteroccocous faecium multirresistente se inicia tratamiento con vancomicina endovenosa, con mejoría clínica. Al ser dada de alta se prescribió como profiláctico trimetoprin sulfametoxasol y fluconazol, con excelente evolución clínica, actualmente asintomática.
DISCUSION
El síndrome de Hiper IgE es una inmunodeficiencia primaria congénita de origen desconocido, cuya fisiopatología permanece aún sin definir. Su distribución en la población es amplia sin predilección por zonas geográficas, grupos étnicos, sexo o condición socioeconómica. La mayor parte de los pacientes no presentan familiares afectos con el mismo síndrome pero estudios recientes describen algunas familias afectas, en las cuales se ha observado un defecto genético en el brazo largo del cromosoma 45. La forma de expresión de esta entidad sugiere un tipo de herencia autosómica dominante con penetrancia variable. En análisis reciente de más de 30 pacientes con síndrome de Hiper IgE y de sus familiares ha permitido definirlo como una enfermedad multisistemica que afecta la dentición, el tejido conectivo y sistema inmune7. Se manifiesta en las primeras semanas de vida, con una susceptibilidad notable a las infecciones bacterianas recurrentes localizadas en piel, aparato respiratorio y óseo y debidas especialmente a Staphilococus aureus8 con desarrollo de neumonías y neumatoceles. También presentan susceptibilidad a infecciones fúngicas que incluyen moniliasis oral y onicomicosis por Candida albicans9. Otro hallazgo característico en estos enfermos es la presencia de dermatitis crónica, pruriginosa, evidente ya en las primeras semanas de vida. Sus características y distribución difieren de la dermatitis atópica, observándose mayor compromiso en la cara, axilas, ingles y periné y superficies de extensión describiéndose poca base eritematosa. A diferencia de los pacientes con dermatitis atópica, en el síndrome de Hiper IgE suele faltar el antecedente familiar de atopia y las infecciones cutáneas no son solamente superficiales sino que afectan a tejido celular subcutáneo7. El retraso en la caída de la dentición primaria, es otro rasgo clínico que puede aparecer y ayudar al diagnóstico, observable hasta en un 60 %-72 % de los pacientes10.
Al llegar a la adolescencia es característico que los pacientes presenten unos rasgos faciales distintivos: asimetría facial que recuerda a una hemi-hipertrofia, frente prominente, ojos hundidos, puente nasal ancho, punta nasal carnosa y prognatismo leve11. Se asocia con anormalidades del sistema en esquelético en rangos diferentes como se observa en otras inmunodeficiencias. Grimbacher et al describen las siguientes en una serie de 30 pacientes, osteoporosis de curso temprano, con fracturas anormalmente frecuentes (hasta en un 57 %) e hiper-laxitud articular y escoliosis hasta en un 63 %5. No hay una constante de laboratorio que asegure el diagnóstico, pero existen diferentes alteraciones, que junto a los hallazgos clínicos permiten establecerlo.
El dato más significativo es la cifra de IgE sérica total, con valores generalmente superiores a 2.000 UI/L. Es posible niveles elevados de IgD, hasta en el 63 % de los pacientes12. Las concentraciones séricas de una o más de las restantes inmunoglobulinas también pueden estar aumentadas, pero generalmente se encuentran valores normales.
Se ha observado defecto en la quimiotaxis de los neutrófilos, pero no es un hallazgo constante y puede variar en un mismo paciente a lo largo del tiempo8. El tratamiento es de soporte y profiláctico para las infecciones en piel con antibióticos tópicos o antimicóticos2. Este Síndrome continúa siendo un misterio inmunológico y ha de definirse por la existencia de criterios clínicos compatibles con dermatitis, eccemas, abscesos en piel, candidiasis, infecciones pulmonares por Staphylococus, características faciales y dentales, alteraciones esqueléticas y niveles elevados de IgE > 2.000 UI/ml.
El diagnostico se estableció por clínica sugestiva de dermatitis, infecciones (otomastoiditis, candidiasis crónica, celulitis), doble arcada dental, IgE muy elevada, aumento en la Ig D, hipergamaglobulinemia y exclusión de: hipersensibilidad, inmunodeficiencia humoral y celular, deficiencia de moléculas LAD I y LAD II.
