


Long-term follow up of patients with hyper IgE syndrome (HIES), as a primary immunodeficiency disorder, has been poorly investigated. This study describes common clinical and immunological features of patients with HIES in the last 10 years in Shiraz University of Medical Sciences, Shiraz, Iran.
Methods and patientsIn this cross-sectional study, the symptoms and medical records of 18 patients, who were diagnosed with HIES, were observed. Genetic and immunologic study was also performed.
ResultsEighteen patients with the mean age of 13 years old were investigated. Ten patients were detected to have mutations in DOCK8 gene and autosomal recessive HIES (AR-HIES); and four patients were found with STAT3 mutation and autosomal dominant HIES (AD-HIES). So, 14 patients with known genetic results were considered for further data analysis. Food allergy, eczema, viral and skin infections were the major complications of AR-HIES patients. The major clinical complications of AD-HIES patients were pneumonia, skin infections and eczema. Food allergy and viral infection were significantly higher in DOCK8 deficient patients. The most common causes of hospitalization in both AR-HIES and AD-HIES patients were pneumonia, skin infections and sepsis. The most common cause of death was found to be sepsis.
ConclusionsAD-HIES and AR-HIES cannot be differentiated only based on the clinical presentations. Genetic features are also necessary for better diagnosis. This study, summarizing the clinical, immunological and genetic information of the patients with AD-HIES and AR-HIES, may open a way for better diagnosis and management of HIES.
Hyper IgE syndrome (HIES) is a rare primary immunodeficiency disorder, involving both humoral and cellular immune systems. This syndrome was previously introduced in separate studies as Job's and Buckley's syndromes.1,2 Following further investigations, it was revealed that these two syndromes describe the same disease which was, then, named as autosomal dominant HIES (AD-HIES).3 Another type of HIES has also been described as autosomal recessive HIES (AR-HIES).
AR-HIES and AD-HIES have some characteristics in common such as recurrent skin infections, elevated serum IgE level, respiratory tract infections and eczema. However, some features are distinct to AD-HIES, such as skeletal, dental and connective tissue abnormalities and some are specific for AR-HIES, such as more susceptibility to viral infections, severe allergy and neurologic complications.4 These two types of the disease also differ in gene mutations involved in HIES pathology. Dominant negative mutations in signal transducer and activator of transcription-3 gene (STAT3, chromosome 17, MIM=147,060) were identified in the autosomal dominant form of the disease, whereas gene mutations in dedicator of cytokinesis-8 gene (DOCK8, chromosome 9, MIM=243,700) were found to be responsible for AR-HIES.5 Defects in STAT3 and DOCK8 can affect several pathways of the immune system; however, the exact mechanism has not been, exactly, identified yet.
Regarding previous surveys, the approximate number of patients with HIES was about 200 cases,3 distributed among different ethnic groups. According to different prognosis and treatment in AR-HIES and AD-HIES, that bone marrow transplant is possible in AR-HIES, the differentiation of these two genetic types is important.6 Symptoms of the patients might vary over time, so differentiation of AR-HIES and AD-HIES is difficult based on the clinical presentations alone. There are some studies focused on both AD-HIES7 and AR-HIES6,8 cases from different areas of the world. However, to our knowledge, no study can be found which has reported the HIES cases in long-term follow up. Thus, in this study, we aimed to report a ten-year follow up of 18 Iranian HIES patients referred to the immunology tertiary center of Shiraz University of Medical Sciences, Shiraz, Iran.
Methods and patientsA cross-sectional study was carried out on 18 patients with HIES diagnosis, referred to the immunology tertiary center of Shiraz University of Medical Sciences, Shiraz, Iran; from 2006 to 2016.
Data gatheringMedical records of the patients were reviewed and patients were followed. Their demographic information, clinical manifestations and genetic defects were gathered in prepared data gathering sheets.
Inclusion criteriaThe diagnosis of HIES was made clinically by medical history, documented medical information, physical examination and laboratory data. Clinical manifestations were defined as follows: recurrent sinopulmonary infection, pneumatocele, eczema, newborn rash, skin infections, severe infections (osteomyelitis, complicated sinusitis and otitis), characteristic face, scoliosis and skeleton infection or fracture, delayed shedding of the primary teeth, cancers (lymphoma), elevation in serum IgE level (>1000IU/mL), eosinophilia and candidiasis. Furthermore, to confirm the diagnosis of AD-HIES, the national institute of health (NIH) scoring system was applied.9
Exclusion criteriaPatients whose information was compatible with other syndromes (represented with elevated IgE, rash and recurrent infection) such as Netherton-Camel, Omenn and Wiskott–Aldrich syndromes; and severe atopic dermatitis were identified and excluded from the study.
Genetic studyIn order to determine the genetic basis of HIES, patients were evaluated for STAT3 and DOCK8 mutations. The ones without genetic defects in these two genes were, subsequently, evaluated for gene mutations in whole exons sequence.
Statistical analysisData was entered into SPSS 19 for 14 patients with defined genetic results. Descriptive analysis was applied for reporting variables. Moreover, Chi-square test was used for comparing qualitative variables between two types of disease. In all studies, P-value <0.05 was considered to be significant.
ResultsGenetic resultsTen patients (56%) were diagnosed as AR-HIES with DOCK8 mutation (five of these patients were from two families). In addition, genetic analysis revealed four (22%) patients with STAT3 mutation representing AD type of the disease. However, genetic results for another four patients have still been obscure (supplementary Table 1).
Patients’ demographic informationEighteen patients (12 males and six females) with the mean age of 13 years old (ranges from three to 24 years old) were evaluated in this study.
The mean age of AR-HIES patients (five males and five females) was 12.25 years old and six patients were from consanguine families. Two families had multiple affected siblings (with DOCK8 mutations); one was with consanguinity (two affected children, P6 and P7), while the other was without consanguinity (three affected children, P8–P10). Moreover, three families reported to have positive history of young children's sudden death.
In AD-HIES patients (four males), the observed mean age was 16 years old and consanguinity was reported in two families. One of the families reported the positive history of children's sudden death.
More detailed information on demographic and consanguinity of the patients is provided in Online Resource, supplementary Table 1.
Clinical manifestations and HIES scoresBased on the HIES scoring system,9 three of the AD-HIES patients were noted to have NIH HIES scores of 30–40 and one of them was observed with the score of 58. More detailed information on patients’ scores is provided in Online Resource, supplementary Table 1.
The mean age of the first presentations was estimated to be one year old and 3.72 years old for AR-HIES and AD-HIES patients, respectively. The mean age of disease diagnosis was about 7.3 years old and 9.6 years old for AR-HIES and AD-HIES patients, respectively.
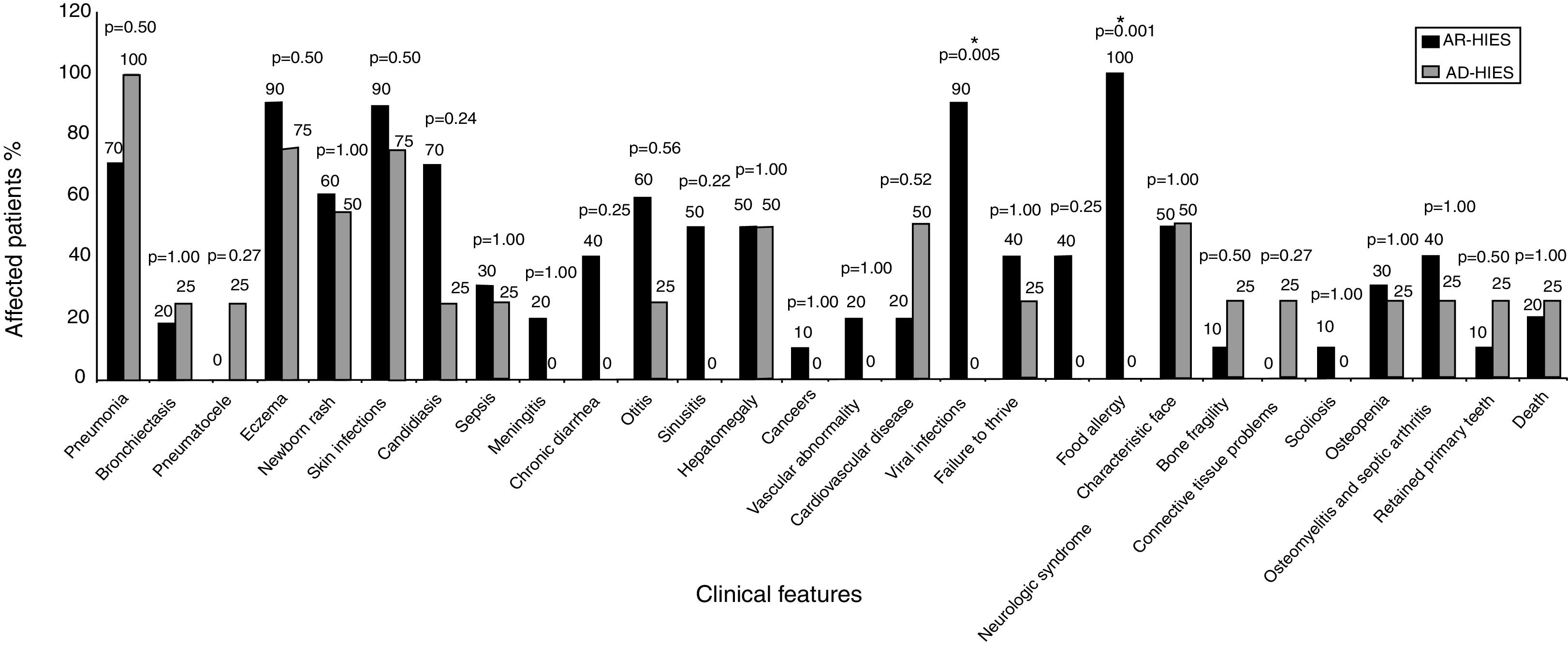
Clinical manifestations of 14 patients were compared between two types of the disease (AD-HIES and AR-HIES), characterized by genetic results, as shown in Fig. 1. Food allergy, eczema, viral and skin infections were the most prevalent clinical manifestations in AR-HIES patients. Pneumonia, skin infections and eczema were highly prevalent in AD-HIES patients.

Comparison of the clinical features of AR-HIES and AD-HIES patients. P-values are represented above each group of clinical features (Fisher's exact test). P-value <0.05 was considered as significant, denoted by *. Vascular abnormality represents vasculitis; and cardiovascular diseases represents myocardial infarction, deep vein thrombosis, high blood pressure.
Clinical presentations of the patients with unknown genetic defect are presented in Table 3 of supplementary data.
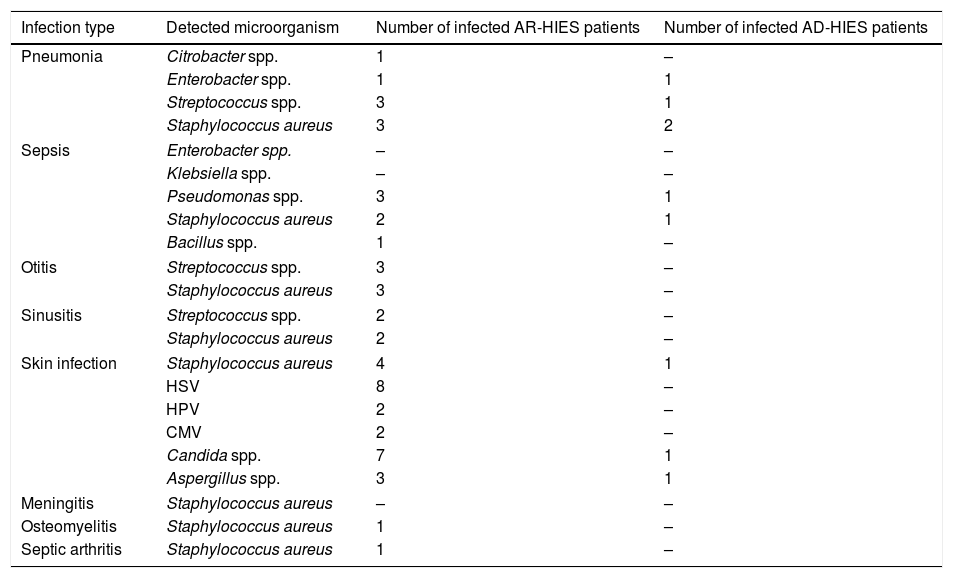
MicroorganismsDetected microorganisms in infections, which were induced following this disorder, are summarized in Table 1.
Microorganisms involved in the infection development in AR-HIES and AD-HIES patients. Bacterial, viral and fungal species are included.
| Infection type | Detected microorganism | Number of infected AR-HIES patients | Number of infected AD-HIES patients |
|---|---|---|---|
| Pneumonia | Citrobacter spp. | 1 | – |
| Enterobacter spp. | 1 | 1 | |
| Streptococcus spp. | 3 | 1 | |
| Staphylococcus aureus | 3 | 2 | |
| Sepsis | Enterobacter spp. | – | – |
| Klebsiella spp. | – | – | |
| Pseudomonas spp. | 3 | 1 | |
| Staphylococcus aureus | 2 | 1 | |
| Bacillus spp. | 1 | – | |
| Otitis | Streptococcus spp. | 3 | – |
| Staphylococcus aureus | 3 | – | |
| Sinusitis | Streptococcus spp. | 2 | – |
| Staphylococcus aureus | 2 | – | |
| Skin infection | Staphylococcus aureus | 4 | 1 |
| HSV | 8 | – | |
| HPV | 2 | – | |
| CMV | 2 | – | |
| Candida spp. | 7 | 1 | |
| Aspergillus spp. | 3 | 1 | |
| Meningitis | Staphylococcus aureus | – | – |
| Osteomyelitis | Staphylococcus aureus | 1 | – |
| Septic arthritis | Staphylococcus aureus | 1 | – |
Some of the patients have experienced several episodes of infections with various microorganisms. Various infections might be observed due to a specific microorganism. The microorganisms involved in some infections were not detected.
CMV, Cytomegalovirus; HPV, Human papilloma virus; HSV, Herpes simplex virus.
The IgE level was markedly elevated in all patients (114–7000IU/mL). The mean IgE level was 2469IU/mL for AR-HIES. Two of the patients had IgE levels less than 1000, two patients were observed with IgE levels between 1000 and 2000 and six patients with IgE levels more than 2000IU/mL. The mean level of IgE was 1501IU/mL for AD-HIES patients. Three patients were noted with IgE levels between 1000 and 2000 and one patient with IgE levels above 2000IU/mL.
Eosinophilia (eosinophil counts more than 450cells/μL) was documented in all of AR-HIES patients with mean levels of 8910cells/μL; and in three of the AD-HIES patients with the mean level of 5866cells/μL.
In this study, two patients (one AR-HIES and one AD-HIES) presented with reduced levels of IgM and one patient (AD-HIES) with low levels of IgG; yet, IgA level was within the normal range in all patients.
B cell deficiency (described as levels less than 5% in flow-cytometry) was reported in two AR-HIES patients. Also, decreased levels of CD3+ cells were observed in two AR-HIES cases. Moreover, decreased levels of CD4+ cells were observed in four AR-HIES and one AD-HIES patient. The levels of CD8+ cells were normal in all patients; however, the reverse CD4+/CD8+ was reported in four AR-HIES and one AD-HIES patients.
More detailed information on immunologic information of the patients is provided in Online Resource, supplementary Table 2.
Hospitalization and mortalityThe most common causes of hospitalization were found to be, respectively, pneumonia, skin infections, HSV infection and sepsis. Pneumonia, skin infections and sepsis were observed in both AD-HIES and AR-HIES; and viral infections were common in AR-HIES.
Two deaths were reported in AR-HIES patients. One was due to Hodgkin lymphoma at age of 4.5 years old and the other was due to sepsis at ages of three. One of the AD-HIES patients died at the age of 13 due to sepsis. Another death was reported due to sepsis in one of the patients with undefined genetic defect.
Treatment and prophylaxisAlmost all patients, following diagnosis of HIES, received antibiotics (mainly cotrimoxazole, co-amoxiclave and azithromycin) as well as antifungals as prophylaxis. Immunoglobulin substitution was also applied for 56% of the patients who were considered to have severe infections.
Two patients required lung lobectomy for preventing further complications. One of the AR-HIES patients received hematopoietic cell transplantation (HCT), which, primarily, got complicated by graft versus host disease (GVHD); however, this complication was resolved and the patient has been well in the past three years.
DiscussionIn this study, we provided the clinical and immunological information of the patients with both AD-HIES and AR-HIES; they were followed for ten years in Shiraz University of Medical Sciences, Shiraz in the south of Iran, with the highest number of diagnosed HIES cases in the country.10
Consanguinity, a predisposing factor for autosomal recessive disorders, is highly prevalent in the south of Iran because of the cultural background in this region.11–13 In the present study, consanguinity was documented in five AR-HIES families (out of seven families) and two AD-HIES families (out of four families). As can be seen in our study, patients from two families were revealed to be AR-HIES without any consanguinity. However, in a previous study by Aydin et al., 99 out of 136 (73%) patients who were diagnosed with AR-HIES were all reported to have consanguine families.6 According to the NIH scoring system, patients with scores <20 are unlikely and ones with scores >40 are highly likely to have AD-HIES. The scores in the range of 20–40 represent the intermediate probability of AD-HIES or might represent another genetic defect of HIES.3 Unlike the mentioned findings, in our study, three AD-HIES were observed with the scores of 20–40. Five AR-HIES patients were also found to have scores ≥40. This can be attributed to variations in different ethnicities and timely dependent presentations of some clinical manifestation of the disease.
The major clinical features of HIES have been mentioned to be eczema, pneumonia, skin abscess and infection, respectively. Other specific symptoms include: skeletal and connective tissue problems, pneumatocele formation and deep abscess, which are mostly present in AD type of the disease. On the other hand, viral infection, neurologic problems, food allergy and asthma are frequently observed in AR type.14
The observed clinical manifestations in AR-HIES and AD-HIES are presented in Fig. 1. In this study, the incidence of pneumonia in AD-HIES patients was relatively lower compared with other studies which have reported recurrent pneumonia in 87% of the AD-HIES patients.14,15 Other studies reported pneumonia in 55% of AR-HIES patients.6 Aydin et al. reported bronchiectasis and pneumatocele in 44% and 3% of the AR-HIES patients, respectively,6 while in this study, only 20% of the AR-HIES patients have experienced bronchiectasis and there were no reports on pneumatocele formation. Pneumatocele and bronchiectasis were both observed in 25% of the AD-HIES patients, which is lower compared to previous studies reporting pneumatocele and bronchiectasis in 52% and 65% of AD-HIES patients, respectively.7
Based on previous surveys, eczema has been the most frequent symptoms of the AD-HIES patients (99–100%).15 However, in our study, pneumonia, eczema and skin infections were reported as the most frequently recorded symptoms of AD-HIES. Skin infections are mostly observed as a result of Staphylococcus aureus in HIES patients and might occur following dermatitis or manifest as skin abscess.
Neonatal rash was also reported to be the first clinical manifestation of the disease. In our study the prevalence of neonatal rash, as the first presentation, was lower than previous reports.14,15 These differences in clinical manifestations might be related to different ethnicity.
Food allergy is one of the frequently observed complications in AR-HIES: 71% of the AR-HIES patients have been reported to suffer from allergies.6 In this study, all of the AR-HIES patients were noted with food allergies that may be due to different food regimen and genetic profile. Food allergy can be described based on a theory which declared that in HIES patients, T-cell response to antigens is preserved, while there is impaired response to mitogens which might be due to Th2 bias.16–18 Impaired IFN-γ production and impaired IFN-γ/IL10 ratio, involved in Th1 cellular immunity, has been observed in HIES patients which results in infection susceptibility. IL4 and IL10, Th2 pathway cytokines, are involved in humoral immunity and result in elevated IgE and eosinophil; and allergic reactions. High levels of IL4 are observed in HIES patients.17,18
Comparing the clinical manifestations of the two types of the disease, our study revealed that viral invasive infections and food allergy are found only in AR-HIES patients. Other studies also showed that viral infection, (especially HSV) and food allergy, respectively as a non-specific and specific sign of AR-HIES, were significantly higher in AR type of the disease.4 Although, sinusitis, meningitis, chronic diarrhea and failure to thrive were higher in AR-HIES patients compared to AD-HIES ones, the difference was not statistically significant, which is compatible with previous findings.
Distinct cases were also reported in our study. As an example, one patient with DOCK8 mutation, surprisingly, filled both AR-HIES and AD-HIES phenotype criteria; including fractures with minor trauma, scoliosis, osteopenia along with HSV infection, food allergy, neurologic syndrome (brain atrophy, seizure and ataxia), sepsis, cellulitis, candidiasis and sub retinal fibrosis. These observations might be affected by differences in genetic defects among different ethnic groups.
A wide range of microorganisms were suggested to cause infectious diseases in HIES patients. Among them, Staphylococcus aureus is considered to be the main cause of skin infections and Streptococcus pneumonia as well as Haemophilus influenza is shown to have a major role in pulmonary infections in AD-HIES.15 In contrast with previous findings, Streptococcus pneumonia as well as Haemophilus influenza was not frequently observed in pulmonary or other sites of infections in our patients. Different laboratory tests and exposure conditions might explain incompatibility. In this study, in AD-HIES patients Staphylococcus aureus was observed predominantly. HSV and Candida species were the most prevalent causes of infections in AR-HIES patients (Table 1). According to previous studies, low Th17 counts and impaired IL-17 production are major pathologic factors which lead to infection by Candida albicans. The rate of this organism in infectious diseases of HIES patients was previously estimated to be 83% in AD-HIES and 92% in AR-HIES patients.14,15 Nevertheless, in our study, it was reported to be lower especially in AD-HIES, which might be due to ethnic differences.
The elevated IgE level is one of the immunological hallmarks of HIES which resulted from impaired IL-12/IFN-ɣ pathway. Borges, et al. demonstrated that in these patients the production of IFN-ɣ (stimulated by IL-12), as an IgE down regulator, is suppressed.19 Serum IgE levels >1000IU/mL were noted in 80% of AR-HIES and 100% of AD-HIES patients. IgE levels >2000IU/mL in 60% of AR-HIES and 25% of AD-HIES patients were also detected in our study. Deficiencies of serum immunoglobulins were not reported previously in HIES patients except for IgM, which was found to show lower levels in AR-HIES patients.5,20 Our study detected low levels of IgM in two (one AD-HIES and one AR-HIES) patients as well as low levels of IgG in one patient (AD-HIES). Differences in our findings with previous reports might be due to this fact that patients might suffer from more than one immunodeficiency disease or immune deviations, during the time. Eosinophilia was observed in 100% of our AR-HIES and 75% of AD-HIES patients, which is a little bit lower than previous studies (96% for AR-HIES7 and 90–93% for AD-HIES patients14,15).
Our results on CD4+ and CD8+ cell counts in HIES patients demonstrated that only CD4+ cell counts reduced in HIES patients (four AR-HIES and one AD-HIES). This is compatible with the study by Engelhardt et al.21; and in contrary to the one by Szczawinska-Poplonyk et al.5 which showed that both CD4+ and CD8+ cell levels can be affected in HIES patients.5,21 Further research can provide more information and new insights.
Genetic mutations in STAT3 and DOCK8 have been reported as the main underlying gene defect in most of the patients with AD-HIES and AR-HIES, respectively. STAT3 plays major roles in the signal transduction of various cytokines, wound healing, cancer and angiogenesis; consequently, its genetic defects bring about several consequences such as lack of inflammation in cold abscesses and sever recurrent infections which may lead to pneumatocele formation.22,23 Mutations in DOCK8 may also affect cellular and humeral immunity, due to their role in preserving normal function of CD8 and forming new B memory cells.6,24 Ten of our patients had DOCK8 mutation with AR phenotype and four patients had STAT3 mutation and AD phenotype. Yet, there remained four patients with unknown gene defect; whole exome sequencing may reveal new genes. Although two of them represented the clinical phenotype of AD-HIES, the genetic results did not support mutation in STAT3 gene.
In the study by Freeman et al.,25 severe infections such as cystic infections of pulmonary by Pseudomonas spp. and Aspergillus spp. have been found as the main causes of death in AD-HIES patients. It mandates the role of prophylaxis for fungi and gram-negative bacteria especially in patients with pneumatocele. In the present study, one death in AD-HIES was reported following infection. Infections were also considered as the main reason of death in AR-HIES patients6 and in our study; sepsis was the reason of death in one of the AR-HIES patients. Lymphoma has been reported as another cause of death in both AR-HIES and AD-HIES patients,26 compatible with our findings which reported one death due to lymphoma in AR-HIES. Since the number of deaths was few in this study, the speculation of the main reason and comparison between AR-HIES and AD-HIES is not possible.
To sum up, the present study summarized both clinical and genetic information of patients diagnosed with AR-HIES and AD-HIES, during a long-term follow up in a geographic region with the highest rate of consanguine families. However, there are some limitations, such as the number of patients, especially in the AD type of the disease, which was inevitable due to the rarity of the disease in Iran and other parts of the world. Moreover, considering the previous studies which have introduced HCT to be successful in treating and increasing the survival of patients with the AR type of the disease, our study reported one patient underwent HCT. This may not be possible for other patients due to the lack of appropriate donor.27
Clinical and immunological history of the patients are key factors in early diagnosis of HIES. Moreover, gene sequencing can also be helpful in determining the type of the disease in case the clinical features are not enough for definite diagnosis. Besides, genetic analysis is essential to determine the therapy, as HCT is highly recommended for Dock8 deficiency. The information provided here can be helpful in more accurate diagnosis of the disease. However, further investigations on undefined genetic defects and mechanisms can shed more light on a better understanding and management of the disease.
Funding sourceThis work was supported by Shiraz University of Medical Sciences, Shiraz, Iran.
Conflicts of interestThe authors have no conflict of interest to declare.
The following is the supplementary data to this article:
