






Asthma is a chronic inflammatory airway disease, the incidence of which has increased recently. In order to identify the potential biomarkers for allergic asthma therapy, microarray data were analysed to find meaningful information.
MethodsMicroarray data GSE18965 were downloaded from Gene Expression Ominibus (GEO), including seven asthmatic epithelium samples from children with allergic asthma and nine healthy controls. Limma package was used to detect differentially expressed genes (DEGs) and the criteria were |log fold change|>0.5 and p value<0.05. We used Database for Annotation, Visualization and Integrated Discovery (DAVID) tool to perform GO function and KEGG pathway analysis. STRING database was used to construct protein–protein interaction (PPI) network. MicroRNA (miRNA) regulation network was constructed according to miRecords database.
ResultsWe identified 274 DEGs in asthma epithelium samples comparing with healthy controls. There were 123 up-regulated DEGs and 151 down-regulated DEGs. PPI network analysis showed that TSPO, G6PD and TXN had higher degree. miRNA regulation network demonstrated that miR-16 and miR-15a had higher degree. The target genes of miRNAs were significantly enriched in the apoptosis function.
ConclusionsTSPO, G6PD and TXN, miR-16, miR-15a and apoptosis may be used as the targets for children's allergic asthma therapy.
Asthma is a common chronic inflammatory airway disease, which is characterised by variable airflow obstruction, hyperresponsiveness, airway inflammation and reversibility either spontaneously or as a result of treatment.1 Asthma is triggered by virus and pollutants through exposure of airway epithelial cell (AEC). The damage of AEC results in increased epithelial permeability and penetration of allergens and injurious materials, reflecting an inadequate repair response, an increased susceptibility to injury, or a combination of both.2 There have been a total of 334 million individuals suffering from asthma worldwide. Asthma is the most common chronic disease for children, and death rates range from 0.0 to 0.7 per 100000 in children.3,4 Thus, it is necessary to explore the mechanism of asthma to achieve effective and satisfactory control of asthma in children.
Asthma is characterised by airway hyperreactivity, which is mediated by Th2 lymphocytes producing interleukin-4 (IL-4), IL-5, IL-9 and IL-13.5 T cell- and eosinophil-induced apoptosis has been reported to be an important event in asthma.6 In severe asthma, there is increased cellular proliferation in the airway contributing to a thickened epithelium and lamina reticularis.7 Microarray profiling has been widely used for the analysis of expressed genes associated with asthma. Woodruff et al. found that calcium-activated Cl-channel-1 and plasminogen activator inhibitor-2 were up-regulated in asthma by genome-wide profiling.8 Fibronectin was the only extracellular matrix component, the expression of which was significantly lower in asthmatic AEC.9
Several reports have reported that microRNAs (miRNAs) played important roles in the pathology of asthma. miRNA is small non-coding RNAs and plays a regulatory role.3 miRNA-221 was differentially expressed in asthmatics and controls, suppression of which could inhibit airway inflammation in asthmatics.10 Toll-like receptor 7 was significantly reduced in severe asthma compared to human controls, which was regulated by miRNAs.11 miRNA-221 and miRNA-485-3p have been reported to be up-regulated in paediatric asthma.12
In this study, differentially expressed genes (DEGs) in the allergic asthma samples comparing with healthy controls were screened out. Then Gene Ontology (GO) function and kyoto encyclopaedia of genes and genomes (KEGG) pathway analysis were performed for the DEGs. Subsequently, we constructed protein–protein interaction (PPI) network of these DEGs. Besides, regulating network of interactions between miRNAs and DEGS was constructed according to the information in the miRecords dataset.
Materials and methodsMicroarray dataGene expression profiling GSE18965 of airway epithelial cells was downloaded from Gene Expression Ominibus (GEO) including seven allergic asthma samples and nine healthy controls from children. The annotation platform was GPL96 [HG-U133A] Affymetrix Human Genome U133A Array.
Affymetrix microarray CEL files were converted into expression profile matrix by Robust Multi-array Average (RMA) algorithm of affy package in the R language.13 Probes were mapped to gene name according to annotation information in the platform Affymetrix by Bioconductor software.14 Expression values of multiple probes that correspond to one gene were averaged.
DEGs screening and function analysisDEGs were screened by linear models for microarray data (limma) package in R language.15 The criteria were |logFC|>0.5 (FC: fold change) and p<0.05. Database for Annotation, Visualization and Integrated Discovery (DAVID) online tool was used for GO and KEGG pathway analysis with p<0.05.16–18
PPI network analysisSTRING online database was used to construct PPI network of DEGs.19 The hub genes were screened according to the degree of node. The degree represents the number of interaction partners and was calculated by Perl code.20 Then the cytoscape was used to construct sub-network.21 Then DAVID tool was used to perform function analysis for the sub-network module.
Regulation relation between miRNA and DEGsThe information of miRNA was extracted from miRecords22 and then the miRNA regulation network was constructed. The interactions of miRNAs and targets genes have been experimentally validated previously. Finally, target genes of miRNAs performed GO function analysis.
ResultsDEGs in allergic asthma samples comparing with healthy controlsA total of 12495 DEGs were obtained after normalisation. Then, 274 DEGs were identified in the asthma samples comparing with healthy controls. Among them, 123 DEGs were up-regulated and 151 genes were down-regulated.
GO function and KEGG pathway analysisWe categorised DEGs by known or proposed gene function (Table 1 listed the top 19 categories). These up-regulated genes participated in the regulation of transcription, negative regulation of nucleic acid and nitrogen compound metabolic process; were involved in the cellular components that are intracellular non-membrane-bounded organelle, non-membrane-bounded organelle, and cytosol; and were involved in molecular functions that are RNA combination, structure of ribosome and myosin light streptokinase activity, as well as pathways that may be specifically related to regulation of actin cytoskeleton regulation and ribosomes. Down-regulated genes participated in the response of the organic substance, protein localisation and intracellular transport; were involved in the cellular components such as plasma membrane and cytosol; and were involved in molecular functions such as peptidase activity, endopeptidase activity and actin binding activity, as well as pathways that may be specifically related to metabolism of lysosomes, glutathione and galactose.
GO and KEGG enrichment analysis for differentially expressed genes (DEGs). The top 10 BP (biological process), MF (molecular function), cellular component (CC) terms with DEGs which were significantly enriched were listed.
| Genes | Category | Term | Count | p value |
|---|---|---|---|---|
| Up-regulate genes | GOTERM_BP_FAT | GO: 0045449∼regulation of transcription | 28 | 0.021779 |
| GOTERM_BP_FAT | GO: 0006350∼transcription | 24 | 0.019963 | |
| GOTERM_BP_FAT | GO: 0045934∼negative regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 9 | 0.028924 | |
| GOTERM_BP_FAT | GO: 0051172∼negative regulation of nitrogen compound metabolic process | 9 | 0.031005 | |
| GOTERM_BP_FAT | GO: 0051253∼negative regulation of RNA metabolic process | 8 | 0.014452 | |
| GOTERM_BP_FAT | GO: 0016481∼negative regulation of transcription | 8 | 0.044573 | |
| GOTERM_BP_FAT | GO: 0043009∼chordate embryonic development | 7 | 0.030307 | |
| GOTERM_BP_FAT | GO: 0009792∼embryonic development ending in birth or egg hatching | 7 | 0.031476 | |
| GOTERM_BP_FAT | GO: 0045892∼negative regulation of transcription, DNA-dependent | 7 | 0.04096 | |
| GOTERM_BP_FAT | GO: 0030036∼actin cytoskeleton organisation | 6 | 0.02232 | |
| GOTERM_CC_FAT | GO: 0043232∼intracellular non-membrane-bounded organelle | 26 | 0.009712 | |
| GOTERM_CC_FAT | GO: 0043228∼non-membrane-bounded organelle | 26 | 0.009712 | |
| GOTERM_CC_FAT | GO: 0005829∼cytosol | 16 | 0.01356 | |
| GOTERM_CC_FAT | GO: 0005794∼Golgi apparatus | 12 | 0.016346 | |
| GOTERM_CC_FAT | GO: 0030529∼ribonucleoprotein complex | 8 | 0.037376 | |
| GOTERM_CC_FAT | GO: 0044445∼cytosolic part | 6 | 0.00234 | |
| GOTERM_CC_FAT | GO: 0005840∼ribosome | 6 | 0.010051 | |
| GOTERM_CC_FAT | GO: 0033279∼ribosomal subunit | 5 | 0.007734 | |
| GOTERM_CC_FAT | GO: 0015934∼large ribosomal subunit | 4 | 0.007914 | |
| GOTERM_CC_FAT | GO: 0022626∼cytosolic ribosome | 4 | 0.013265 | |
| GOTERM_MF_FAT | GO: 0003723∼RNA binding | 11 | 0.028442 | |
| GOTERM_MF_FAT | GO: 0003735∼structural constituent of ribosome | 5 | 0.030331 | |
| GOTERM_MF_FAT | GO: 0004687∼myosin light chain kinase activity | 2 | 0.020882 | |
| KEGG_PATHWAY | hsa04810: regulation of actin cytoskeleton | 6 | 0.027998 | |
| KEGG_PATHWAY | hsa03010: ribosome | 4 | 0.032319 | |
| Down-regulate gene | GOTERM_BP_FAT | GO: 0010033∼response to organic substance | 17 | 0.00137 |
| GOTERM_BP_FAT | GO: 0008104∼protein localisation | 17 | 0.009684 | |
| GOTERM_BP_FAT | GO: 0046907∼intracellular transport | 15 | 0.00396 | |
| GOTERM_BP_FAT | GO: 0055114∼oxidation reduction | 14 | 0.007952 | |
| GOTERM_BP_FAT | GO: 0006955∼immune response | 14 | 0.014456 | |
| GOTERM_BP_FAT | GO: 0042592∼homeostatic process | 14 | 0.026908 | |
| GOTERM_BP_FAT | GO: 0045184∼establishment of protein localisation | 14 | 0.031774 | |
| GOTERM_BP_FAT | GO: 0006091∼generation of precursor metabolites and energy | 12 | 2.01E-04 | |
| GOTERM_BP_FAT | GO: 0009719∼response to endogenous stimulus | 11 | 0.005385 | |
| GOTERM_BP_FAT | GO: 0009725∼response to hormone stimulus | 10 | 0.008521 | |
| GOTERM_CC_FAT | GO: 0005886∼plasma membrane | 53 | 0.017878 | |
| GOTERM_CC_FAT | GO: 0044459∼plasma membrane part | 35 | 0.013535 | |
| GOTERM_CC_FAT | GO: 0005829∼cytosol | 26 | 0.003322 | |
| GOTERM_CC_FAT | GO: 0000267∼cell fraction | 22 | 0.0051 | |
| GOTERM_CC_FAT | GO: 0005739∼mitochondrion | 22 | 0.005318 | |
| GOTERM_CC_FAT | GO: 0031226∼intrinsic to plasma membrane | 21 | 0.032332 | |
| GOTERM_CC_FAT | GO: 0005887∼integral to plasma membrane | 20 | 0.046866 | |
| GOTERM_CC_FAT | GO: 0005783∼endoplasmic reticulum | 19 | 0.013103 | |
| GOTERM_CC_FAT | GO: 0005773∼vacuole | 18 | 1.37E−09 | |
| GOTERM_CC_FAT | GO: 0005794∼Golgi apparatus | 18 | 0.011081 | |
| GOTERM_MF_FAT | GO: 0070011∼peptidase activity, acting on l-amino acid peptides | 12 | 0.011274 | |
| GOTERM_MF_FAT | GO: 0008233∼peptidase activity | 12 | 0.015298 | |
| GOTERM_MF_FAT | GO: 0004175∼endopeptidase activity | 9 | 0.021001 | |
| GOTERM_MF_FAT | GO: 0003779∼actin binding | 8 | 0.029168 | |
| GOTERM_MF_FAT | GO: 0048037∼cofactor binding | 7 | 0.026057 | |
| GOTERM_MF_FAT | GO: 0050662∼coenzyme binding | 6 | 0.024665 | |
| GOTERM_MF_FAT | GO: 0003924∼GTPase activity | 6 | 0.043348 | |
| GOTERM_MF_FAT | GO: 0050661∼NADP or NADPH binding | 4 | 0.0042 | |
| GOTERM_MF_FAT | GO: 0051015∼actin filament binding | 4 | 0.012327 | |
| GOTERM_MF_FAT | GO: 0015078∼hydrogen ion transmembrane transporter activity | 4 | 0.048639 | |
| KEGG_PATHWAY | hsa04142: lysosome | 13 | 4.32E−08 | |
| KEGG_PATHWAY | hsa00480: glutathione metabolism | 6 | 5.61E−04 | |
| KEGG_PATHWAY | hsa00052: galactose metabolism | 5 | 3.92E−04 | |
| KEGG_PATHWAY | hsa00030: pentose phosphate pathway | 4 | 0.004625 | |
| KEGG_PATHWAY | hsa00520: amino sugar and nucleotide sugar metabolism | 4 | 0.022122 | |
| KEGG_PATHWAY | hsa00010: glycolysis/gluconeogenesis | 4 | 0.048967 | |
| KEGG_PATHWAY | hsa00410: beta-alanine metabolism | 3 | 0.03614 | |
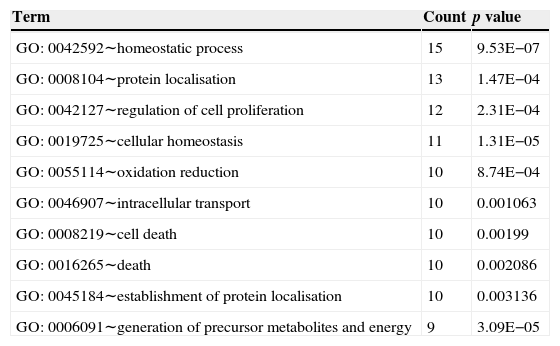
Fig. 1 showed the PPI network of DEGs, demonstrating that TSPO (translocator protein), G6PD (glucose-6-phosphate dehydrogenase deficiency) and TXN (thioredoxin) have the higher degrees which were 38, 20 and 17, respectively. The interactions among these three nodes and their first adjacent nodes were used to construct the sub-network (Fig. 2). The major biology processes which the sub-network modules participated in were homeostatic process, protein localisation and regulation of cell proliferation (Table 2).
")
Protein–protein interaction network which the differentially expressed genes corresponded to. Red dots represent up-regulated genes and green dots represent down-regulated genes. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of the article.)
")
Sub-network of top three hub proteins and their first adjacent nodes of hub proteins. Red dots represent up-regulated genes and green dots represent down-regulated genes. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of the article.)
Top 10 biology processes in which genes in the sub-network were significantly enriched.
| Term | Count | p value |
|---|---|---|
| GO: 0042592∼homeostatic process | 15 | 9.53E−07 |
| GO: 0008104∼protein localisation | 13 | 1.47E−04 |
| GO: 0042127∼regulation of cell proliferation | 12 | 2.31E−04 |
| GO: 0019725∼cellular homeostasis | 11 | 1.31E−05 |
| GO: 0055114∼oxidation reduction | 10 | 8.74E−04 |
| GO: 0046907∼intracellular transport | 10 | 0.001063 |
| GO: 0008219∼cell death | 10 | 0.00199 |
| GO: 0016265∼death | 10 | 0.002086 |
| GO: 0045184∼establishment of protein localisation | 10 | 0.003136 |
| GO: 0006091∼generation of precursor metabolites and energy | 9 | 3.09E−05 |
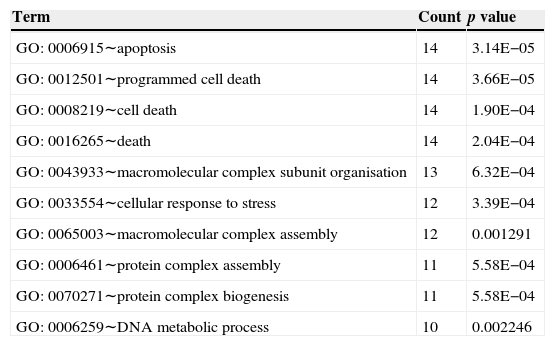
Fig. 3 shows the relation network between miRNA and DEGs. In the network, miR-16 and miR-15a have the higher degrees, which were 62 and 59, respectively. The target genes of miRNA participated in the cell apoptosis, programmed cell death and cell death (Table 3).
. Yellow dots represent miRNAs and green dots represent DEGs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of the article.)")
Top 10 biology processes in which the targets genes of miRNA were enriched.
| Term | Count | p value |
|---|---|---|
| GO: 0006915∼apoptosis | 14 | 3.14E−05 |
| GO: 0012501∼programmed cell death | 14 | 3.66E−05 |
| GO: 0008219∼cell death | 14 | 1.90E−04 |
| GO: 0016265∼death | 14 | 2.04E−04 |
| GO: 0043933∼macromolecular complex subunit organisation | 13 | 6.32E−04 |
| GO: 0033554∼cellular response to stress | 12 | 3.39E−04 |
| GO: 0065003∼macromolecular complex assembly | 12 | 0.001291 |
| GO: 0006461∼protein complex assembly | 11 | 5.58E−04 |
| GO: 0070271∼protein complex biogenesis | 11 | 5.58E−04 |
| GO: 0006259∼DNA metabolic process | 10 | 0.002246 |
In this study, we identified 274 DEGs in the asthma samples comparing with healthy controls. Then we used PPI network to identify hub genes and screened key miRNAs which may be associated with asthma.
PPI network analysis showed that TSPO, G6PD and TXN had the high degree. TSPO is an 18kDa protein mainly found in the mitochondrial membrane,23 which has been shown to alter mitochondrial respiration in a dose dependent manner, increasing the rate of succinate-supported state IV respiration and decreasing ADP-supported state III respiration, thereby decreasing the respiratory control ratio.24 The severity of atopic dermatitis, a chronic inflammatory skin disease, was related with TSPO expression in male patients.25 It has been reported that TSPO is highly expressed on the bronchial and bronchiole epithelium in the lungs of humans and guinea pigs,26 pneumocytes and alveolar macrophages27 in humans. However, there were no reports about the relation between TSPO and asthma. In this study, TSPO had a high degree in the PPI network. The hub protein which had a high degree tends to be essential.28 Therefore, we speculated that TSPO might be a potential biomarker for asthma therapy and provided basic understanding for the pathologic understanding of asthma. G6PD is a cytosolic enzyme that catalyses the chemical reaction from d-glucose 6-phosphate to 6-phospho-d-glucono-1,5-lactone.29 In individual embryos, mRNAs expression of G6PD was correlated with respiration rates and uptake of oxygen.30 The expression of antioxidant gene G6PD was induced in the enhanced severity of the asthmatic mice in response to allergen through RT-PCR analysis.31 In acute exacerbations of asthma, the serum TRX levels in patients with asthma were significantly increased compared to those during the asymptomatic period.32 There was a high sensitisation rate of thioredoxin among bakers.33 In ovalbumin-sensitised mice, TRX strongly suppressed airway hyperresponsiveness and airway inflammation.34 Thus, G6PD and TRX may be biomarkers in the airway epithelial cells for asthma therapy.
The sub-network of hub proteins (TSPO, G6PD and TRX) was significantly enriched in the homeostatic process. Surfactant homeostasis was altered in the Aspergillus fumigatus-induced allergic airway inflammation, demonstrating the down-regulation of hydrophobic surfactant proteins (SPs), SP-B and SP-C and increase in SP-D protein levels.35 Altered l-arginine homeostasis and deficiency of nitric oxide due to increased arginase activity have been considered to be the causative factors in the pathology of asthma.36 Thus, the homeostasis process may be one of the important biological processes in the pathogenesis of allergic asthma.
Interaction network of miRNA and DEGs showed that miR-16 and miR-15a had high degree. Lower levels of miR-15a decreased the expression of vascular endothelial growth factor-A which is over-expressed in the sputum and serum of both adult and paediatric patients with asthma.37 miR-16 was up-regulated in the mice when exposing the airways to house dust mite by quantitative PCR analysis.38 The miR-15 and miR-16 in the network demonstrated their relation with the DEGs in the asthma tissue. We could conclude that miR-15 and miR-16 might regulate the genes in the asthma tissue in response to stimulus. The target genes of miRNAs were significantly enriched in apoptosis, programmed cell death, and cell death. Asthma is characterised by the accumulation of eosinophils into the lungs, which participate in the maintenance and exacerbation of inflammation. Inflamed airway eosinophil apoptosis has been observed by anti-asthmatic agent such as glucocorticoids, theophylline and leukotriene modifiers, which may be a therapeutic target in the treatment of allergic asthma.39 Besides, it has been reported that the spontaneous and mitogen-induced apoptosis of mononuclear cells were increased in patients with atopic asthma.40 Thus, apoptosis, also called programmed cell death, may be the potential target for allergic asthma.
In summary, TSPO, G6PD and TXN with a high degree in the PPI network may be potential targets for asthma therapy. In addition, miR-15 and miR-16 might be the important miRNAs which regulate the DEGs in allergic asthma. The apoptosis pathway could also be the target for asthma therapy. Lack of experiments is the drawback of our study. So the experiments for investigating the roles of TSPO, G6PD, TXN, miR-15 and miR-16 in the allergic asthma would be needed in a future study.
Conflict of interestThe authors declare that they have no conflict of interests to state.
Authors’ contributionsY.Q.C. carried out the design and coordinated the study, participated in most of the experiments and prepared the manuscript. J.N.Q. provided assistance in the design of the study, coordinated and carried out all the experiments and participated in manuscript preparation. All authors have read and approved the content of the manuscript.
FundingThe National Natural Science Fund Project (No. 81170028).
Ethical disclosuresPatients’ data protectionConfidentiality of data. The authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
