


Stevens-Johnson syndrome (SJS) is a life-threatening vesiculobullous disease characterized by an acute eruption that involves the skin and mucous membranes. Various etiologic factors have been implicated as a cause of SJS, including infection, vaccination, drugs, systemic diseases, physical agents, and food. Drugs are the most commonly blamed.
The incidence of SJS is estimated to be between 1.1 and 7.1 cases per million person-years.1 SJS is currently considered to be a part of bullous disease syndromes [SJS, SJS- toxic epidermal necrolysis (TEN) overlap syndrome, and TEN] in which keratinocyte cell death results in subepidermal separation. In SJS, skin detachment is limited to less than 10% of the body surface area (BSA). TEN requires skin detachment of more than 30% of the BSA. An overlap group of SJS/TEN has been defined with erosions between 10% and 30% of the BSA.1–3 The pathogenesis of SJS has yet to be clarified.
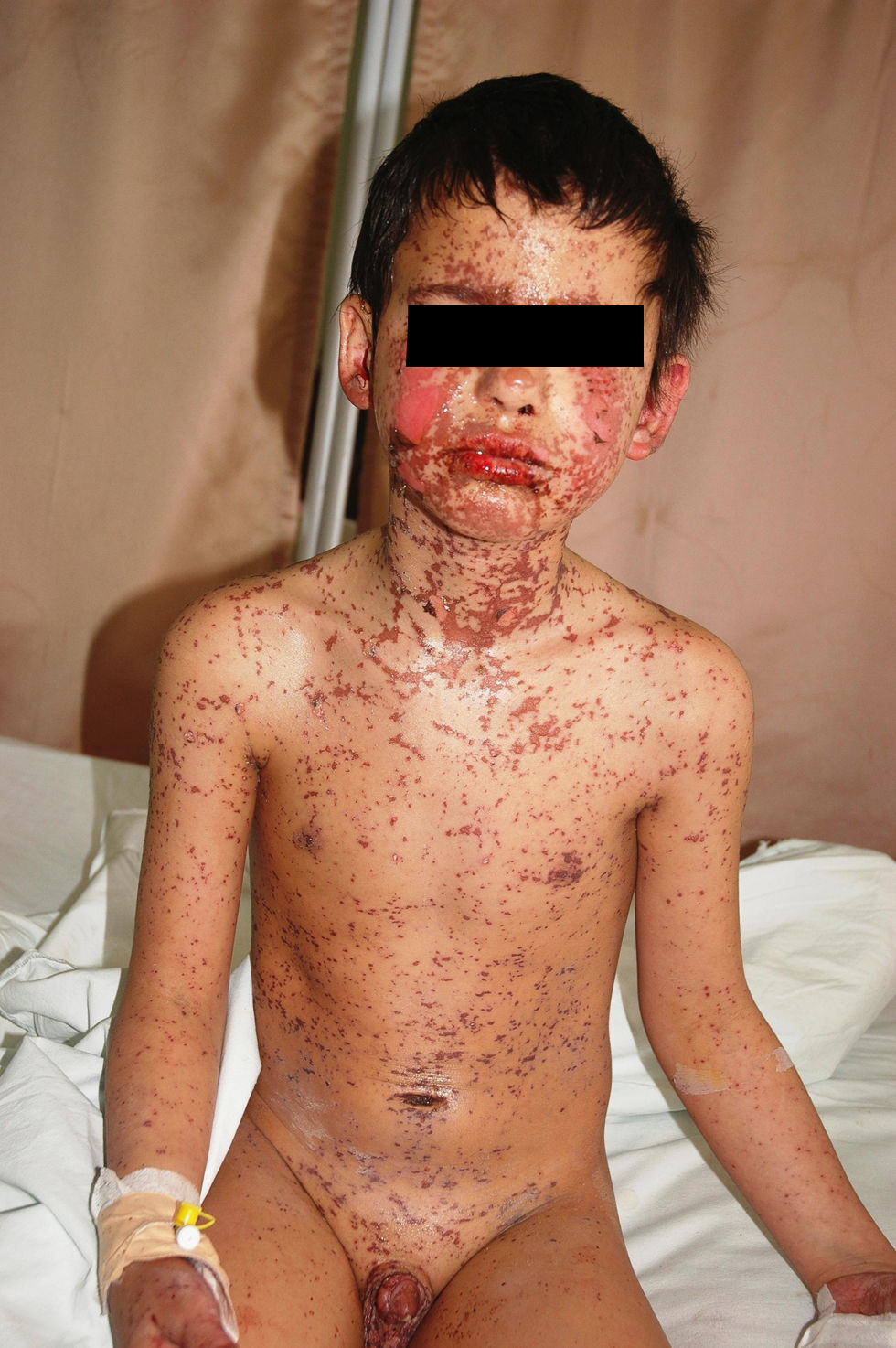
We report a 10-year-old male patient who was admitted to our department with a widespread bullous, erythematous pruritic eruption, bilateral conjunctivitis, oedema of lips and eyelids, haemorrhagic crusts of the lips, superficial erosions of the hard palate, an ulceration on the penile meatus (Fig. 1), and high fever (38.2°C). The symptoms first started 2 days before, approximately 7h after intramuscular administration of the 2nd dose of cefazolin (2×500mg, i.m) which had been prescribed with the diagnosis of acute pharyngitis. He did not take any other drug apart from the antibiotics. He had no history of adverse drug reactions and had not been treated with cefazolin before. Other personal and family history was unremarkable. The clinical diagnosis of drug induced SJS was made with epidermal detachment of 7–8% of total body surface area.1–3

Laboratory results on admission revealed WBC count, 2330/mm3; lymphocytes, 25%; platelets, 160000/mm3; erythrocyte sedimentation rate (ESR), 36/h; C-reactive protein, 5.08mg/dl (0-1); aspartate aminotransferase (AST), 54U/l (15-41); alanine aminotransferase (ALT), 16U/l (14-54). Blood urea nitrogen (BUN), 41mg/dl; creatinin and urine analysis were normal. Serum Na, 131mEq/l; K, 4.6mEq/l; total protein, 5.7gr/dl; albumin, 2.6gr/dl.
Haemoculture and swab culture from skin lesions were negative. Serum cold haemagglutinin titers and viral serology were negative.
With immunophenotypic analysis (Coulter Epics XL), absolute numbers of peripheral blood lymphocytes were calculated as: CD19+, 136; CD4+, 156; CD8+, 236 (CD4+/CD8+ ratio, 0.66), and CD3−CD16+ CD56+ (NK cells), 29cells/μl. The predominant phenotype of the gated lymphocytes among blister fluid mononuclear cells was CD8+ (48.7%). Other cell subsets were CD19+ (0.2%), CD4+ (25.2%), and NK cells (3.4%).
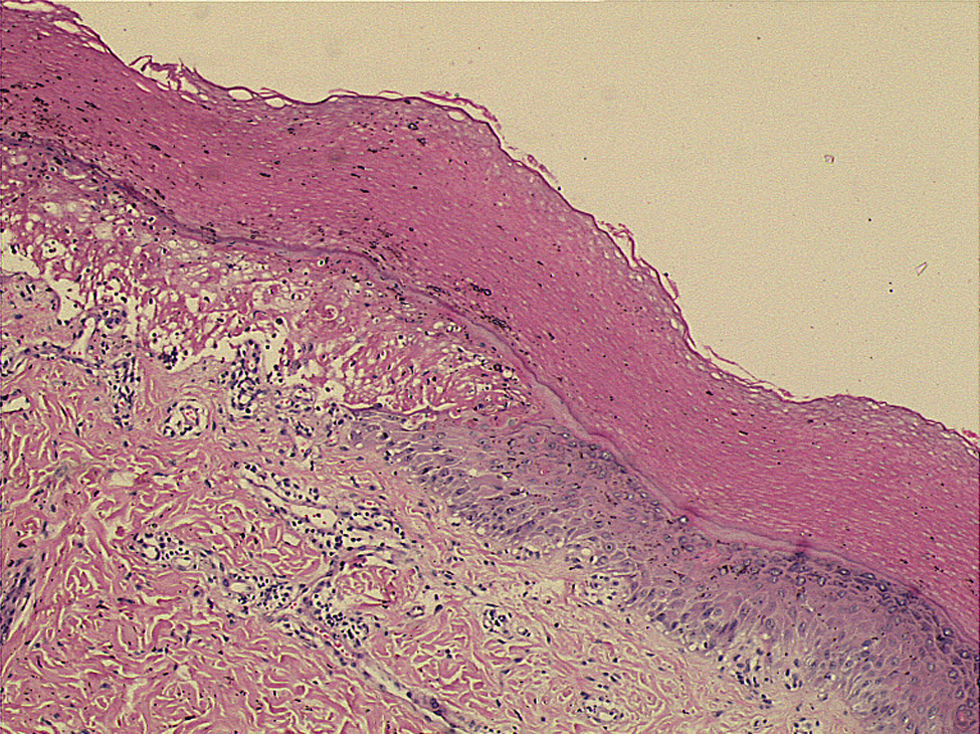
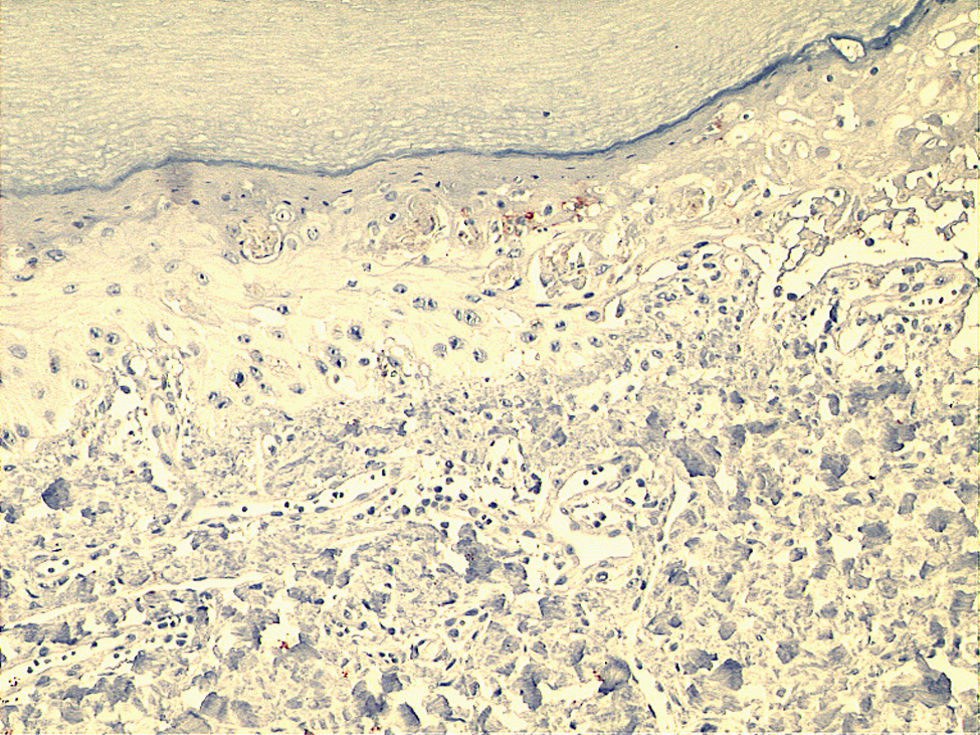
Microscopically, the surface epithelium of the skin biopsy was hyperkeratotic. There was full-thickness epithelial necrosis, dermo-epidermal separation, as well as sparse lymphocyte exocytosis into the epithelium, and sparse perivascular leukocyte infiltration of the upper dermis (Fig. 2). The epidermal lymphocytes showed labelling with anti-CD8 by immunohistochemical staining. These changes were considered to be consistent with SJS (Fig. 3).

Squamous epithelium on skin surface is hyperkeratotic. Note full-thickness epithelial necrosis and dermo-epidermal separation on the left corner as well as sparse lymphocyte exocytosis into the epithelium and sparse perivascular leukocyte infiltration of the upper dermis. H&E, X50.

The patient's symptoms began to improve after initiation of intravenous methylprednisolone and hydroxizine in conjunction with supportive therapy. Corticosteroid dose was gradually tapered after the 4th day of treatment. He had gastrointestinal bleeding during his hospitalisation. This is attributed to the gastrointestinal system involvement in SJS. The patient was discharged with markedly clinical improvement 12 days after admission. Follow-up visits were conducted by our Ophthalmology Clinic for his dry eye syndrome as a long term effect of SJS. The patient was patch tested with cefazolin 2 months after the reaction. However, he removed the test tape after 24h because of itching and did not return for evaluation.
SJS is an uncommon and potentially serious mucocutaneous disease. More than 100 drugs have been associated with the development of SJS and TEN.4 Cefazolin would be responsible for inducing SJS in our patient. The role of corticosteroid therapy in SJS is still controversial.1,2,5 Our patient's symptoms improved after initiation of intravenous methylprednisolone and hydroxizine in conjunction with supportive therapy.
The pathogenesis of SJS has yet to be clarified. The scenario suggested by today's literature points towards drug-specific CD8+cytotoxic T cells utilising perforin/granzyme B trigger keratinocyte apoptosis.6 In TEN, several studies stated a predominance of CD8+ T lymphocytes along dermoepidermal junction, in the epidermis, or in the blister fluid of early blisters.7–11 In keeping with these findings, Behrendt et al.12 reported a higher percentage of perforin positive CD8+ T lymphocytes in PBMC of patients with SJS on the day of their admission than in healthy donors. Similarly, Psodas et al.13 showed increased mRNA expression of perforin and granzyme B in PBMC of SJS and TEN patients compared to controls and suggested that there may even be association between disease severity and perforin and granzyme B levels. The expression levels were markedly higher in mononuclear cells obtained from blister fluid compared to PBMC, suggesting local production of both of these transcripts.6 These findings lend further support to the theory that keratinocyte apoptosis is triggered by drug-specific cytotoxic T lymphocytes (CTL) using the perforin/granzyme B pathway. Subsequently, there may be an expansion of apoptosis involving the interaction of either membrane-bound or soluble Fas ligand (sFasL) with its receptor Fas. However, the cellular source of sFasL remains controversial, with both peripheral lymphocytes and keratinocytes themselves as potential candidates.2,6
Our data are in agreement with previous reports of the presence of CD8+ T cells at the site of skin lesions in patients with SJS,7–12 and further support the involvement of cytotoxic CD8+T cells in eliciting tissue damage in SJS. We concluded that the cells contained in the blister fluid during SJS would provide an interesting way to investigate the immune mechanisms in bullous drug reactions.
