


Las conectivopatías hereditarias tales como el síndrome de Marfan, síndrome de Ehlers-Danlos, pseudoxantoma elástico y osteogénesis imperfecta, se han asociado con arteriopatía periférica y desarrollo de aneurismas1. El síndrome de Alport es una enfermedad genética de la síntesis de colágeno de tipo IV (membrana basal) manifestado clínica-mente por una nefropatía progresiva, asociada con una pérdida auditiva sensorioneural que afecta de preferencia a frecuencias de entre 4.000 y 8.000 Hz, acalasia y anomalías oculares con cataratas posteriores, luxación del cristalino, distrofia corneal, nistagmo y miopía. Este síndrome se debe a mutaciones del gen del colágeno, con una transmisión semidominante, ligada al sexo en el 85%.
CASO CLÍNICODescribimos el caso de un hombre asiático, de 36 años de edad, que se presentó en el servico de urgencias con dolor constante, intenso, bilateral en la fosa lumbar, que lo despertaba y se asociaba a náuseas y sudación. A los 19 años de edad se le diagnosticó un síndrome de Alport y a los 21 años recibió un trasplante renal de cadáver en el eje ilíaco derecho, antes de lo cual había sido sometido a hemodiálisis durante 5 años. También experimentaba sordera y había requerido cirugía por catarata bilateral. Su medicación habitual incluía: ciclosporina, atenolol, prednisolona y amlodipino. Entre los factores de riesgo de enfermedad cardiovascular destacaba la presencia de tabaquismo (5-10 cigarrillos al día) e hipertensión arterial. No tenía antecedentes de sífilis, traumatismos, tuberculosis o septicemia ni antecedentes familiares de conectivopatía; su padre presentaba una coronariopatía. En el momento del ingreso la exploración reveló una masa pulsátil, dolorosa, de 8 cm de diámetro, en el abdomen asociada con una ligera distensión abdominal. En las investigaciones iniciales se identificó una proteinuria de 3+ y una concentración de creatinina plasmática de 188 mmol/l; el paciente se encontraba estable desde un punto de vista cardiovascular y afebril. Se consideró un diagnóstico de litiasis renal. La ecografía abdominal y la tomografía computarizada revelaron la rotura contenida de un aneurisma aórtico toracoabdominal tipo IV, de 10 cm de diámetro, que afectaba a la arteria mesentérica superior y al eje celíaco (fig. 1). Antes de la reparación del aneurisma, se implantó un injerto de derivación axilobifemoral para el riego del riñón trasplantado y ambas piernas. En el postoperatorio, el paciente requirió 3 días de hemofiltración antes de que los niveles de creatinina empezaran a normalizarse. En el octavo día postoperatorio se produjo la oclusión del injerto axilobifemoral. Fue reingresado 3 semanas después del alta con un absceso en la anastomosis axilar y el injerto se extrajo sin incidentes.
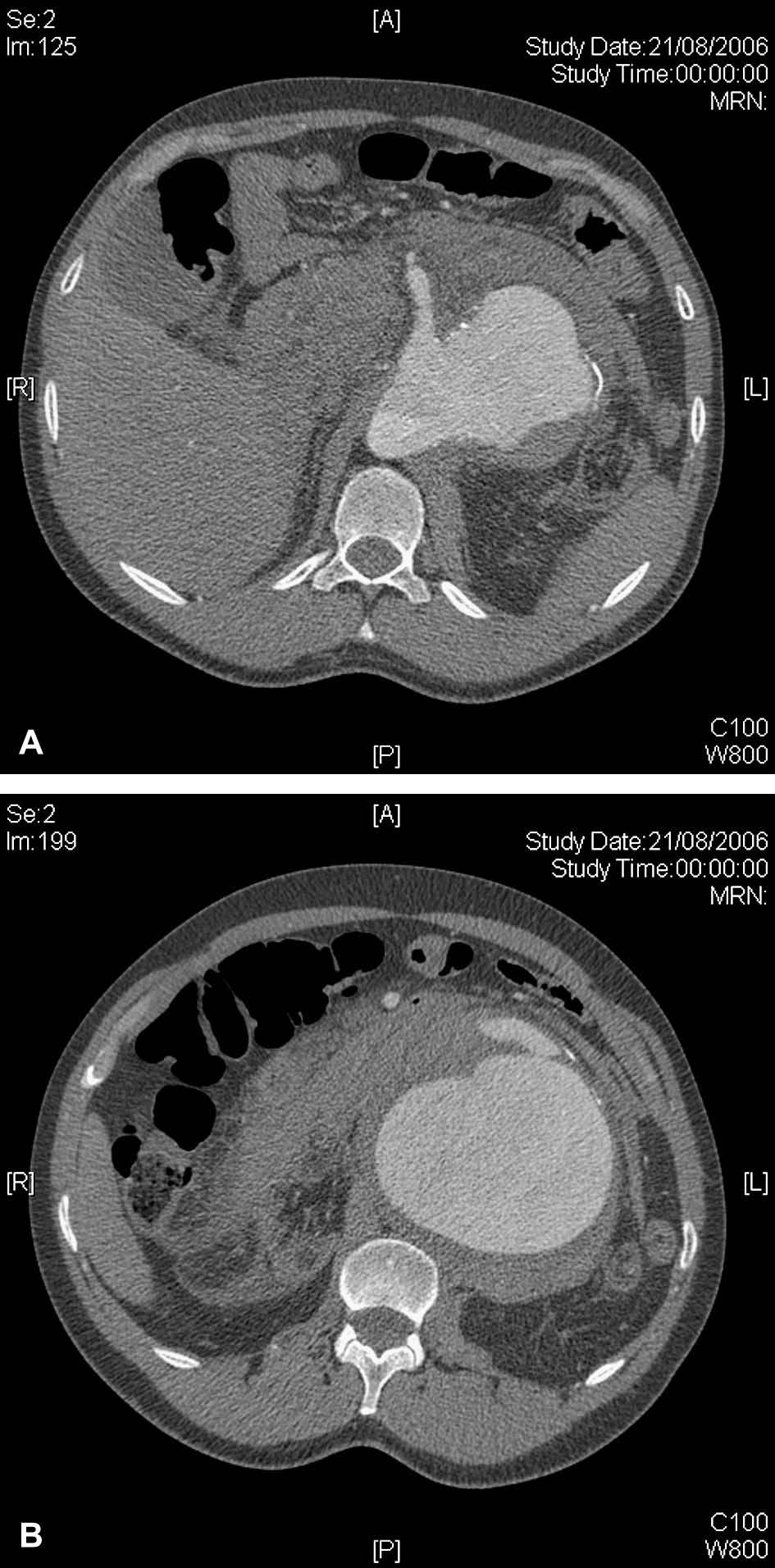
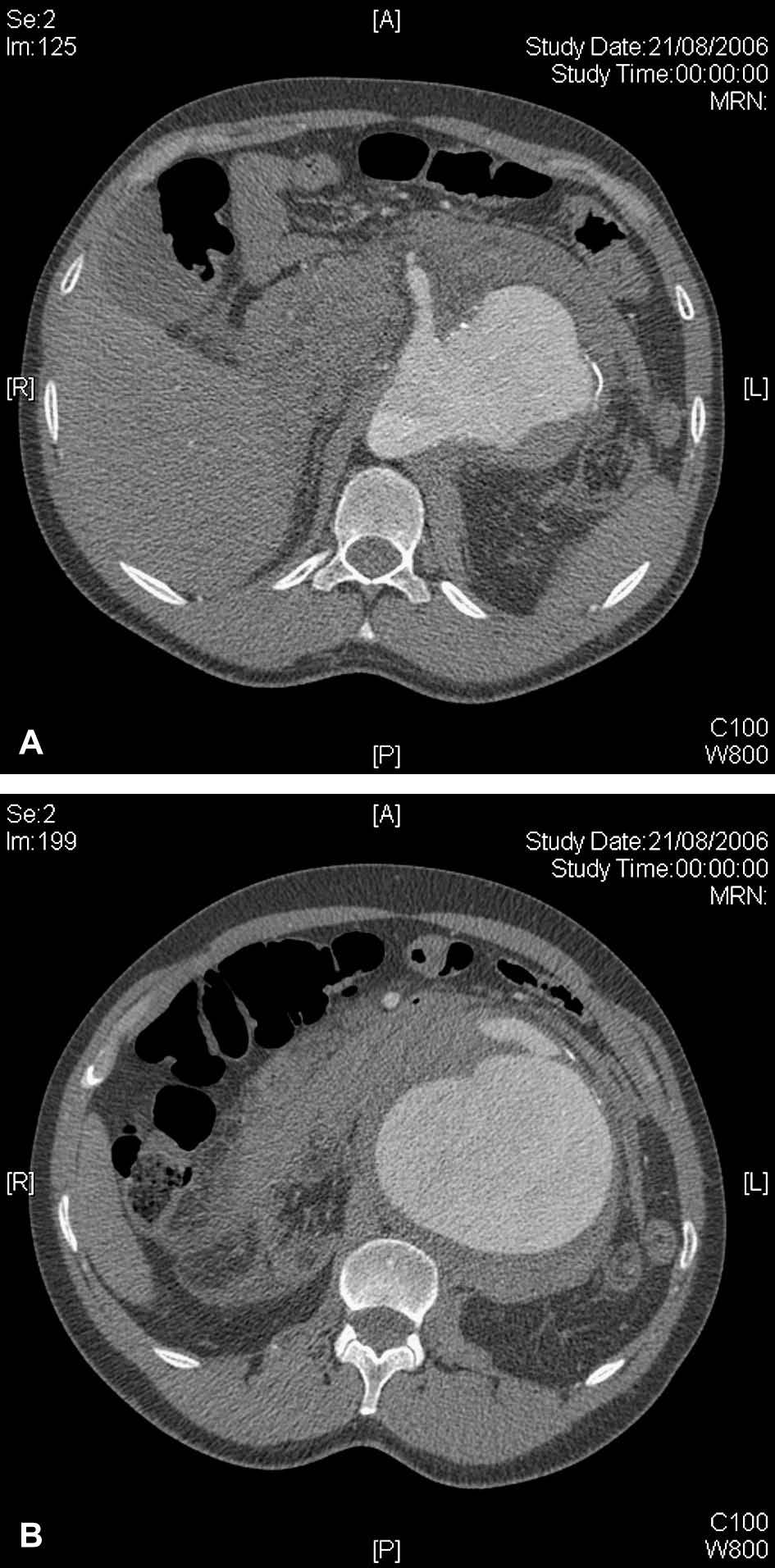
Fig 1. A. Tomografía computarizada (TC) que revela un aneurisma aórtico toracoabdominal tipo IV que afecta a la arteria mesentérica superior. B. TC axial que pone de manifiesto la rotura contenida de un aneurisma aórtico toracoabdominal tipo IV de 10 cm de diámetro.
DISCUSIÓNLa asociación entre el síndrome de Alport y una vasculopatía es previsible, pero es sorprendente que se hayan publicado tan pocos casos, dado que muchas conectivopatías hereditarias, en particular del colágeno tipo III y IV, se han implicado en la patogenia de los aneurismas2. Una búsqueda actual de los estudios publicados en todo el mundo ha identificado un caso clínico de un aneurisma intracraneal en un paciente joven con síndrome de Alport3.
Es bien conocido que los aneurismas aórticos abdominales (AAA) se asocian a edad avanzada, sexo masculino, tabaquismo, aterosclerosis, hipertensión arterial y predisposición genética4. Además, ee ha demostrado que la diálisis crónica da lugar a una aceleración de la aterosclerosis y se ha asociado con una mayor incidencia de muerte súbita provocada por la disección de aneurismas aórticos5. Tanto la hipertensión arterial como la aterosclerosis grave se han implicado en este proceso y en general, se considera que los AAA son la manifestación final de la aterosclerosis severa4,6. Siguen sin conocerse los mecanismos celulares que causan la degeneración aneurismática específica de la aterosclerosis. La ciclosporina A, un inmunosupresor inhibidor de la calcinurina, se ha asociado con hipertensión arterial, hiperlipemia, disfunción endotelial y aterosclerosis acelerada. Los mediadores de la disfunción vascular asociada a la ciclosporina A incluyen la actividad simpática excesiva, déficit relativo de óxido nítrico, factor de crecimiento transformante beta 1, endotelina 1, especies reactivas del oxígeno y del nitrógeno y eicosanoides vasoconstrictores7. El tratamiento con este fármaco inmunosupresor se ha identificado como posible factor etiológico en la disección espontánea de la arteria coronaria8. Debido a la edad relativamente joven de este paciente, proponemos una posible asociación entre el síndrome de Alport y el desarrollo de un AAA.
Hasta lo que los autores conocen, el paciente descrito en el presente informe es el primer caso publicado en todo el mundo sobre una asociación entre el síndrome de Alport y un aneurisma aórtico toracoabdominal. A pesar de que no podemos demostrar que el aneurisma se desarrolló como consecuencia del defecto en la síntesis de colágeno tipo IV, los aneurismas aórticos pueden ser más frecuentes en pacientes con este síndrome que en la población sana. Un defecto en el gen del colágeno tipo IV podría constituir una etiología probable En pacientes con el síndrome que presentan lumbalgia aguda, en particular cuando se dispone de pruebas poco firmes de una litiasis renal, es preciso considerar el diagnóstico de rotura de aneurisma aórtico. El caso descrito en el presente informe también destaca la técnica quirúrgica de usar una derivación axilobifemoral antes de clampar la aorta. Los beneficios de esta técnica incluyen una disminución de la isquemia de las extremidades y del riñón trasplantado, la disminución de sobrecarga precarga cardíaca, y su contribución en reducir los cambios de la presión arterial asociados al clampaje de la aorta.
Correspondencia: Peter R. Taylor, MA, MChir, FRCS, Department of Surgery, 1st Floor North Wing, St. Thomas’ Hospital, Lambeth Palace Road, Londres SE1 7EH, Reino Unido. Correo electrónico: taylorvasc@aol.com
Ann Vasc Surg. 2007;21:816-8 DOI: 10.1016/j.avsg.2007.06.004©Annals of Vascular Surgery Inc. Publicado en la red: 13 de agosto de 2007