


Puntos clave
- •
Las anomalías congénitas del riñón y del tracto urinario engloban un amplio grupo de trastornos que se originan por un defecto en el desarrollo del aparato urinario a diferentes niveles.
- •
Puede haber alteración en el parénquima renal, como la displasia multiquística, anomalías en la migración embriológica, como la ectopia renal o el riñón en herradura, y alteraciones en el sistema colector, desde la pelvis (estenosis de la unión pieloureteral) hasta la uretra (válvulas de uretra posterior).
- •
En el riñón displásico, el tejido renal normal está reemplazado por múltiples quistes de diferentes tamaños separados por una cantidad variable de parénquima displásico. Cuando los quistes son muy pequeños y predomina el componente estromal se denomina displasia quística sólida.
- •
La ectopia renal ocurre cuando el riñón no asciende desde la pelvis hacia la fosa renal. Si cruzan la línea media, se denomina ectopia renal cruzada.
- •
La estenosis de la unión pieloureteral (EPU) es la causa más frecuente de obstrucción en el tracto urinario superior.
- •
El megauréter constituye aproximadamente el 20% de los casos de hidronefrosis en el neonato, constituyendo la segunda causa detrás de la EPU. La mayoría son del tipo megauréteres refluyentes no obstructivos.
Las anomalías congénitas del riñón y del tracto urinario (ACRTU) representan aproximadamente el 20-30% de todas las alteraciones identificadas en el periodo prenatal1. Engloban un amplio grupo de trastornos que se originan por un defecto en el desarrollo del aparato urinario en diferentes niveles:
- –
Las malformaciones del parénquima renal aparecen por un fallo en el desarrollo normal de la nefrona y dan como resultado displasia, hipoplasia, agenesia renal (AR), disgenesia tubular renal y enfermedades quísticas.
- –
Anomalías en la migración embriológica del riñón dan lugar a ectopia renal y defectos de fusión como el riñón en herradura.
- –
Alteraciones en el desarrollo del sistema colector conllevan anomalías en la pelvis renal, como la obstrucción de la unión pieloureteral; del uréter, como el megauréter primario, uréter ectópico, ureterocele o reflujo vésico ureteral; en la vejiga, como la extrofia vesical, y en la uretra, como las válvulas de uretra posterior (VUP).
Los defectos pueden ser uni o bilaterales y pueden coexistir varios en un mismo paciente. Son responsables del 30-50% de los casos de enfermedad renal terminal y, por lo tanto, es importante su diagnóstico y tratamiento para prevenir o disminuir el daño renal2 y evitar la progresión a enfermedad renal terminal.
EpidemiologíaLas anomalías congénitas del riñón y del tracto urinario se presentan en 3-6/1.000 nacidos vivos y son responsables del 34-59% de enfermedad renal crónica y del 31% de los casos de enfermedad renal terminal3. Wiesel et al analizaron las anomalías del aparato urinario de 709 nacimientos, incluyendo nacidos vivos, muertos y abortos. De todas ellas, la dilatación del tracto urinario fue la afección más prevalente, detectándose en el 28%4. Aunque muchas formas de ACRTU se presentan asociadas a síndromes con defectos multiorgánicos, la mayoría de los ACRTU ocurren en pacientes no sindrómicos5,6. Los síndromes con ACRTU se desarrollan junto con otras anomalías fuera del riñón y del tracto urinario, y tienen manifestaciones clínicas características de su síndrome, mientras que en las ACRTU no sindrómicas las alteraciones están confinadas únicamente al riñón y al tracto urinario. Hay evidencias basadas en modelos de experimentación animal que apoyan la hipótesis de que el desarrollo de ACRTU en pacientes no sindrómicos está originado por defectos de un solo gen7–9.
Malformaciones del parénquima renalUn fallo en la nefrogénesis puede ocasionar un defecto en el desarrollo renal y puede originar: displasia renal, AR, disgenesia tubular renal, displasia multiquística o enfermedad renal poliquística.
La patogenia de dichas alteraciones es multifactorial y están implicados factores genéticos y ambientales10–15.
- –
Factores genéticos. Hay varios genes relacionados con el desarrollo de alteraciones en el parénquima renal, como los genes EYA1 y SIX1, FRAS1 y PAX2 que originan displasia renal asociada a distintos síndromes. La enfermedad poliquística renal está relacionada con mutaciones en el gen PKD1 en la forma autosómica dominante y el PKHD1 en la autosómica recesiva.
- –
Factores ambientales. La exposición a agentes teratogénicos y las deficiencias de ciertos nutrientes está relacionada con ACRTU. La administración prenatal de inhibidores de la enzima conversiva de angiotensina (IECA) y bloqueadores de los receptores de la angiotensina II está relacionada con hiperplasia yuxtaglomerular14,16. En animales de experimentación, se ha demostrado que la deficiencia de la vitamina A está asociada a malfor maciones genitourinarias y a hipoplasia renal17.
Lectura rápida
Las anomalías congénitas del riñón y del tracto urinario (ACRTU) son una causa frecuente de morbilidad y fallo renal en niños. Representan aproximadamente el 20-30% de todas las alteraciones identificadas en el periodo prenatal. Factores genéticos y ambientales están implicados en el fallo del desarrollo embriológico normal del aparato urinario dando lugar a un amplio grupo de trastornos a diferentes niveles, según donde se altere la embriogénesis:
- –
Las malformaciones del parénquima renal aparecen por un fallo en el desarrollo normal de la nefrona y dan como resultado displasia, hipoplasia, agenesia renal (AR), disgenesia tubular renal y enfermedades quísticas. La hipoplasia es una entidad en la que se observan menor número de cálices y nefronas, pero sin componentes displásicos ni embrionarios, es decir, el riñón es estructuralmente normal a diferencia de la displasia renal. En la displasia multiquística el tejido renal normal está reemplazado por múltiples quistes de diferentes tamaños separados por una cantidad variable de parénquima displásico, mientras que poliquistosis renal es una enfermedad genética progresiva de los riñones con 2 formas de presentación según el patrón de herencia y puede afectar al riñón y a otros órganos.
- –
Anomalías en la migración embriológica del riñón da lugar a ectopia renal y defectos de fusión, como el riñón en herradura. La ectopia renal ocurre cuando el riñón no asciende desde la pelvis hacia la fosa renal (a nivel de la segunda vértebra lumbar). Se sitúan en su lado pero se asientan en una mala posición. En la ectopia renal cruzada el riñón ectópico cruza la línea media y se fusiona con el riñón contralateral, pero el uréter del riñón ectópico mantiene su inserción en la vejiga. El riñón en herradura consiste en 2 masas renales situadas verticalmente en la línea media o a uno de sus lados, unidos por un istmo de parénquima o fibrosis que cruza la línea media.
- –
Alteraciones en el desarrollo del sistema colector conllevan anomalías en la pelvis renal, como la obstrucción de la unión pieloureteral (EPU); del uréter, como el megauréter primario, uréter ectópico, ureterocele o reflujo vesicoureteral; en la vejiga, como la extrofia vesical, y en la uretra, como las VUP. La estenosis de la EPU es la causa más frecuente de obstrucción en el tracto urinario superior. Se puede definir como aquella situación en la que existe una restricción al flujo de orina desde la pelvis renal hacia el uréter, produciendo en consecuencia la dilatación de los cálices y la pelvis renal, y si no es tratada un deterioro renal progresivo. Las anomalías a nivel de la unión ureterovesical pueden dar lugar a las siguientes afecciones: megauréter obstructivo no refluyente, megauréter no obstruido y refluyente, megauréter refluyente y obstructivo, y megauréter no refluyente y no obstructivo.
El megauréter no refluyente y obstructivo primario está producido por una anomalía en la musculatura distal del uréter con una hipoplasia de las fibras musculares y una hipertrofia de las fibras intersticiales de colágeno; todo ello origina un segmento adinámico que causa una obstrucción más o menos significativa.
En algunos casos, también hay asociado un túnel submucoso corto que hace que la orina refluya hacia el uréter y no sea capaz de bajar (megauréter refluyente y obstructivo).
Cuando la orina refluye pero drena sin complicaciones hacia la vejiga, hablamos de un megauréter refluyente no obstructivo, y si no se objetiva ni reflujo ni obstrucción pero se evidencia un uréter dilatado, se trata de un megauréter no refluyente ni obstructivo. Este último pudiera deberse a un megauréter primario obstructivo, con buena evolución y maduración del extremo distal del uréter, dejando de ser obstructivo pero manteniendo la dilatación ureteral.
El ureterocele consiste en una dilatación quística del segmento intravesical del uréter. El 80% se asocia al polo superior de un riñón duplicado y el 60% tiene un orificio ectópico en la uretra.
Los defectos pueden ser uni o bilaterales y pueden coexistir varios en un mismo paciente.
La dilatación del tracto urinario es la afección más prevalente, detectándose hasta en el 28%.
Aunque muchas formas de ACRTU se presentan asociadas a síndromes con defectos multiorgánicos, la mayoría de los ACRTU ocurren en pacientes no sindrómicos.
Son responsables del 30-50% de los casos de enfermedad renal terminal y, por lo tanto, es importante su diagnóstico y tratamiento para prevenir o disminuir el daño renal y evitar la progresión a enfermedad renal terminal.
La hipoplasia es una entidad en la que se observan menor número de cálices y nefronas, pero sin componentes displásicos ni embrionarios, es decir, el riñón es estructuralmente normal, a diferencia de la displasia renal. El diagnóstico de hipoplasia se establece cuando coexisten los siguientes criterios10:
- –
Reducción del tamaño renal 2 desviaciones estándar de la media según la edad.
- –
Exclusión de daño renal con ácido dimercaptosuccínico-Tc 99m (DMSA).
- –
En casos de hipoplasia renal unilateral, hipertrofia compensadora del riñón contralateral.
El riñón displásico es el resultado de una diferenciación metanéfrica anormal, observándose elementos primitivos de forma difusa, focal o segmentaria.
Si aparecen quistes, se denomina displasia quística, y si hay una preponderancia de quistes, displasia multiquística. La aplasia renal puede ser en ocasiones el resultado de un estadio involucionado de riñón multiquístico. Los riñones displásicos pueden ser funcionales o estar anulados. La displasia renal puede ser uni o bilateral y su prevalencia es de 2-4/1.000 nacimientos18. El tamaño suele variar, pero generalmente es menor que el de un riñón normal, aunque suele estar determinado por la presencia o ausencia de quistes. La displasia renal puede detectarse prenatalmente, apreciándose un incremento de la ecogenicidad debido al tejido parenquimatoso anormal, pobre diferenciación corticomedular y quistes parenquimatosos. Pueden presentarse asociados a otras anomalías del tracto urinario, como hidronefrosis, duplicaciones renoureterales, megauréter obstructivo y reflujo vesicoureteral (RVU)11.
Displasia multiquísticaEs importante distinguir entre multiquístico y poliquístico. El riñón multiquístico se refiere a una enfermedad displásica y la poliquistosis renal es una afección que incluye varias enfermedades, todas con nefronas normales y sin displasia.
En la displasia multiquística el tejido renal normal está reemplazado por múltiples quistes de diferentes tamaños, separados por una cantidad variable de parénquima displásico. Estos riñones no son reniformes ni tienen sistema calicial de drenaje. Cuando los quistes son muy pequeños y predomina el componente estromal, se denomina displasia quística sólida (fig. 1).

Su incidencia es de 0,3-1/1.000 nacidos vivos y la mitad de ellos se diagnostican prenatalmente19,20. La mayoría de los casos involucionan, como se demuestra en las ecografías posnatales realizadas de manera periódica. La tasa de involución es más frecuente durante los 2 a 3 primeros años de vida21,22. Es el tipo más frecuente de enfermedad quística renal y de displasia renal en niños. Es la segunda causa en frecuencia de masa abdominal durante el periodo neonatal.
Pueden encontrarse alteraciones del tracto urinario en el riñón contralateral, siendo las más frecuentes la estenosis de la unión pieloureteral y el RVU, que puede encontrarse hasta en el 25% de los casos23.
Se ha descrito asociación a hipertensión y neoplasia renal en algunos casos. Hay publicaciones que muestran restos nefrogénicos en estos riñones en un 4% y en los riñones normales entre un 0,8 y 1%24–26.
Enfermedad genética quísticaPoliquistosis renal autosómica recesivaSe pueden distinguir 2 formas en función de que la enfermedad se presente en el periodo perinatal o durante la infancia. Su prevalencia oscila entre 1/5.000 a 1/40.000 recién nacidos vivos, pero un 50% muere en las primeras horas o días por uremia o fallo respiratorio. La herencia es autosómica recesiva y el gen se aloja en el brazo corto del cromosoma 16.
En los neonatos, los riñones están aumentados de tamaño pero mantienen su forma normal. El riñón presenta dilataciones quísticas fusiformes radiales de la médula a la corteza. El tejido intersticial está moderadamente incrementado. Después del periodo neonatal, los riñones disminuyen de tamaño, los quistes se hacen menos prominentes y la fibrosis intersticial aumenta a medida que lo hace la insuficiencia renal.
Los pacientes tienen cierto grado de fibrosis hepática. Generalmente, si la enfermedad hepática es leve, el riñón se afecta más y viceversa.
Poliquistosis renal autosómica dominanteSe presenta en 1/500 a 1/1.000 recién nacidos y su herencia es autosómica dominante. De forma típica, se presenta en adultos entre 30 y 50 años. El riñón puede llegar a ser enorme por el crecimiento de los quistes. Estos varían en tamaño y, aunque los cálices y la pelvis son normales, pueden estar distorsionados por la compresión ejercida por los quistes. A medida que la enfermedad avanza, el parénquima renal llega a ser sustituido por los quistes. Suelen ser bilaterales.
NefronoptisisEs la forma más frecuente de enfermedad renal quística recesiva. Se caracteriza por la presencia de túbulos renales anormales, inflamación intersticial y fibrosis. Se han identificado distintas mutaciones recesivas, NPHP1-11, NPHP1L, SDCCAG8, relacionadas con esta afección. La presentación suele ser la aparición lenta de fallo renal con síntomas como poliuria, polidipsia y disminución de la capacidad de concentración de la orina.
Agenesia renalLa AR se caracteriza por la ausencia de parénquima renal y se origina por una disrupción en el desarrollo metanéfrico en una fase temprana. Su incidencia es de 1/2.900 nacimientos y la AR unilateral representa el 5% de las malformaciones renales4,27. En su patogenia están implicados múltiples factores, como las mutaciones de genes implicados en el desarrollo renal (Ret o GDnf), y agentes teratogénicos, como el ácido retinoico y la cocaína. Pueden presentarse asociados a otras alteraciones genitourinarias28. La enfermedad con la que más frecuentemente coexiste es el RVU, que se presenta en el 37% de los pacientes con AR unilateral. Otras anomalías asociadas frecuentemente son la estenosis de la unión pieloureteral (6-7%) y la estenosis de la unión ureterovesical (11-18%). Pueden presentar síntomas derivados de la anomalía con la que coexisten. En niñas con AR, hay riesgo de anomalías müllerianas, como el útero bicorne y la duplicación vaginal, entrando dentro del espectro de malformaciones del síndrome de Mayer-Rokitansky.
Antes de diagnosticar un paciente de AR, se debe verificar la ausencia de tejido renal en todo el abdomen y no solo en la fosa renal. Este hecho se demuestra en una revisión de 13.705 fetos seguidos prenatalmente con ecografía29, en la que se identificó a 40 fetos con fosa renal vacía. En la ecografía posnatal se observó que la causa fue en 24 pacientes una ectopia renal, en 13 una AR, en 2 un riñón en herradura y en un caso una ectopia renal cruzada (ERC).
Disgenesia tubular renalSe caracteriza por la ausencia o un escaso desarrollo de los túbulos proximales. La fisiopatología de la enfermedad no está clara, pero se ha observado que se origina en fetos por alguna de estas 3 circunstancias:
- 1.
Entidad autosómica recesiva (OMIM 267430).
- 2.
Exposición a fármacos IECA.
- 3.
En el feto donante en los casos de síndrome de transfusión feto-fetal.
La mayoría de los pacientes mueren en periodo perinatal por la hipoplasia pulmonar y la secuencia Potter.
Anomalías de la migración renalLa migración y la rotación renal se completan en la novena semana de gestación30. Las anomalías renales congénitas en la posición y en la fusión son el resultado de la interrupción de la migración embriológica normal de los riñones31.
Ectopia renalSe estima una incidencia de 1 por cada 1.000 autopsias. Ocurre cuando el riñón no asciende desde la pelvis hacia la fosa renal (a nivel de la segunda vértebra lumbar). Se sitúan en su lado, pero se asientan en una mala posición. En el riñón ectópico hay también una falta de rotación de anterior a medial, situándose por tanto la pelvis más anterior. Si cruzan la línea media se denomina ERC. La ERC puede presentarse con o sin fusión al riñón contralateral. El aporte arterial del riñón ectópico varía y puede provenir de la arteria iliaca, de la aorta y en algunas ocasiones de la arteria hipogástrica o de las arterias sacras.
Se puede presentar asociada a otras malformaciones renales, sobre todo con el RVU. También se asocia a malformaciones de otros órganos (genitales, esqueleto) y formando parte de diversos síndromes polimalformativos32,33.
El riñón ectópico suele tener una función disminuida como se demuestra en una serie de 99 casos de riñones ectópicos en la que se observó una función disminuida en el DMSA en 74de 82 riñones34.
Anomalías de la fusión renalOcurren cuando una porción de un riñón se fusiona con el contralateral. La anomalía de fusión más frecuente es el riñón en herradura.
Riñón en herraduraSu incidencia oscila entre 1/400 y 1/800 recién nacidos vivos, según diferentes series35. Consiste en 2 masas renales situadas verticalmente en la línea media o a uno de sus lados, unidas por un istmo de parénquima o fibrosis que cruza la línea media. En el 90% de los casos están unidos por el polo inferior y generalmente esta unión o istmo está situada en la parte baja del abdomen, por delante de los grandes vasos y debajo de la unión de la arteria mesentérica inferior y la aorta. La fusión se produce antes de que los riñones asciendan desde la pelvis a su posición normal en la fosa renal, generalmente antes de la quinta semana de gestación. Esta fusión implica una falta de rotación, por lo que las pelvis renales se sitúan anteriores. Hay una asociación frecuente con otras anomalías. Las anomalías urológicas asociadas con más frecuencia son el RVU y la hidronefrosis, presentes en el 26-32% y el 8-15%, respectivamente, según distintas series36.
La hidronefrosis puede llegar a ser obstructiva en el 30% de los casos37. Hay autores que demuestran en sus series que la hidronefrosis obstructiva se desarrolla con el tiempo38. Esta particularidad nos hace pensar que los RH deben ser seguidos en su evolución a lo largo del tiempo con ecografías seriadas. El aporte vascular del istmo es proporcionado por un vaso aislado y puede provenir de la aorta, de la iliaca común o de la arteria mesentérica inferior. Un vaso polar que inicialmente no produce obstrucción puede con el crecimiento del riñón llegar a dificultar el paso a través de la unión pieloureteral.
Ectopia renal cruzadaEn este caso, el riñón ectópico cruza la línea media y se fusiona con el riñón contralateral, pero el uréter del riñón ectópico mantiene su inserción en la vejiga. Su incidencia se estima en 1/2.000 nacidos. En la mayoría de los casos, el riñón ectópico se fusiona al polo inferior del riñón contralateral, que puede situarse en su posición dorsolumbar normal o pélvico. Su diagnóstico suele efectuarse con ecografía, pero en ocasiones se requiere otras pruebas de imagen como la urorresonancia magnética (uro-RM) para confirmar el diagnóstico y conocer la anatomía, principalmente si el paciente va a ser intervenido.
Alteraciones en el desarrollo del sistema colectorEstenosis de la unión pieloureteralLa estenosis de la unión pieloureteral (EPU) es la causa más frecuente de obstrucción en el tracto urinario superior. La incidencia de hidronefrosis es del 1-1,4% en recién nacidos, pero la mayoría de los casos se resuelven espontáneamente y la incidencia de EPU baja al 0,2-0,4%39,40.
Actualmente, la mayoría de los casos se diagnostican prenatalmente como hidronefrosis. Si no es detectada intraútero, se puede manifestar en niños como masa abdominal palpable, infección urinaria, hematuria o molestias gastrointestinales. De las masas abdominales en niños menores de un año, el 50% son de origen renal y el 40% de ellas son secundarias a EPU.
Para su diagnóstico se debe efectuar una ecografía. En los casos detectados prenatalmente, esta se realiza a partir de los 3-7 días de vida (cuando comienza la diuresis fisiológica del recién nacido).
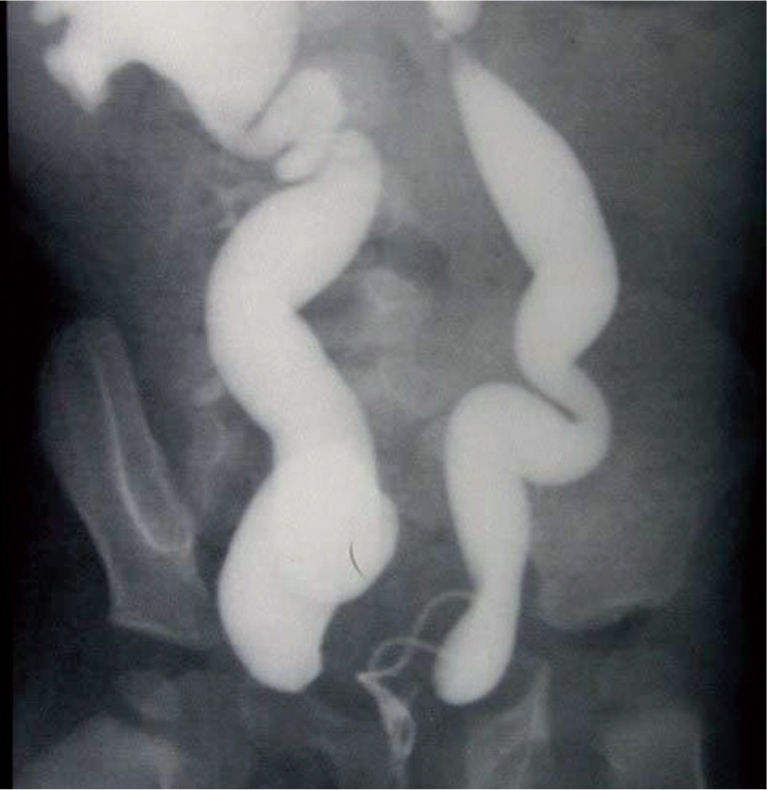
Si en la ecografía se ve dilatación ureteral, realizamos una cistografía miccional seriada (CUMS) para descartar RVU (33% de los casos) (fig. 2).

Si la hidronefrosis es de grado II o inferior, controlamos con ecografías en 6 meses-1 año, y en las hidronefrosis grado III o superior, y si la pelvis es mayor de 15mm, hay que pedir un renograma isotópico diurético. Generalmente, se emplea el isótopo MAG-3 (ácido mercapto-acetil-triglicina).
Según la mayoría de los autores, los pacientes con una hidronefrosis moderada o severa y un patrón de eliminación obstructivo deben ser tratados mediante cirugía41. Optan por esta actitud para preservar la función y evitar el deterioro renal con la obstrucción. Otros autores prefieren intervenir solo aquellos con función renal diferencial menor del 40%, independientemente de que haya un patrón obstructivo de eliminación.
Megauréter congénitoEl término megauréter es un término descriptivo para un uréter dilatado (mayor de 7mm). El megauréter primario se debe a una alteración funcional o anatómica que afecta a la unión ureterovesical. Se puede dividir en megauréter obstructivo no refluyente, megauréter no obstruido y refluyente, megauréter refluyente y obstructivo, y megauréter no refluyente y no obstructivo.
El megauréter no refluyente y obstructivo primario está producido por una anomalía en la musculatura distal del uréter con una hipoplasia de las fibras musculares y una hipertrofia de las fibras intersticiales de colágeno; todo ello origina un segmento adinámico que causa una obstrucción más o menos significativa42. En algunos casos, también hay asociado un túnel submucoso corto que hace que la orina refluya hacia el uréter y no sea capaz de bajar (megauréter refluyente y obstructivo). Cuando la orina refluye pero drena sin complicaciones hacia la vejiga, hablamos de un megauréter refluyente no obstructivo, y si no se objetiva ni reflujo ni obstrucción pero se evidencia un uréter dilatado, se trata de un megauréter no refluyente ni obstructivo. Este último pudiera deberse a un megauréter primario obstructivo con buena evolución y maduración del extremo distal del uréter, dejando de ser obstructivo pero manteniendo la dilatación ureteral.
El megauréter constituye aproximadamente el 20% de los casos de hidronefrosis en el neonato, constituyendo la segunda causa detrás de la EPU. La mayoría son del tipo megauréteres refluyentes no obstructivos. La incidencia de megauréter primario es de 0,36 cada 1.000 nacidos vivos.
Una vez diagnosticado el megauréter, es importante determinar la causa, ya que una parte va a requerir tratamiento para salvaguardar la función renal.
Hay que determinar qué pacientes presentan un megauréter obstructivo, un refluyente o ambos. Para descartar el RVU, efectuamos una CUMS y para diagnosticar el megauréter obstructivo realizamos un renograma isotópico, generalmente con MAG-3. Con el renograma diurético, podemos evaluar la función renal y si existe obstrucción a nivel de la unión ureterovesical.
El megauréter no obstructivo y no refluyente se maneja de manera conservadora, con controles periódicos con ecografía y renograma diurético.
El tratamiento del megauréter obstructivo también debe ser expectante, dada la elevada tendencia a la mejoría y a la resolución espontánea43. Aquellos pacientes con megauréter primario y manejo conservador deben ser tratados de manera profiláctica con antibióticos44. La duda principal es decidir qué pacientes deben ser intervenidos. En general, en aquellos pacientes con función renal diferencial (FRD) conservada (>40%) se opta por el manejo conservador. Si la FRD disminuye por debajo del 40%, si hay un gran dolicomegauréter con codos que generen obstrucción o si se producen infecciones urinarias se debe optar por la intervención.
También se considera la posibilidad de realizar una derivación externa, tipo nefrostomía o ureterostomía, o interna, como un catéter doble J, de manera temporal hasta la intervención para evitar la progresión del daño renal43.
UreteroceleConsiste en una dilatación quística del segmento intravesical del uréter. El 80% se asocia al polo superior de un riñón duplicado y el 60% tiene un orificio ectópico en la uretra.
La manera más frecuente de presentación es como infección urinaria en los primeros meses de vida. También puede ser detectado en las ecografías prenatales. Otra forma menos frecuente de manifestación es la obstrucción de la uretra, aunque en niñas constituye la causa más frecuente de obstrucción uretral.
Se debe realizar CUMS a todos los pacientes porque el 50% del polo inferior ipsolateral y el 25% contralateral tienen RVU. El renograma isotópico diurético nos marcará la función de cada hemirriñón y si existe obstrucción a nivel de la unión ureterovesical.
Uréter ectópicoSe habla de uréter ectópico cuando el orificio ureteral acaba en una posición caudal a su inserción normal en el trígono. Generalmente, se localiza en el trayecto de formación de los conductos mesonéfricos. Eso implica que en niños el meato se puede situar desde la región uretral de los conductos eyaculadores hasta el epidídimo y en las niñas desde el cuello vesical hasta la uretra, vagina o más infrecuente el cérvix o el útero45.
Reflujo vesicoureteralEs el flujo retrógrado anormal de la orina vesical hacia el tracto urinario superior a través de una unión urétero vesical incompetente, de forma primaria con base genética o de forma secundaria por obstrucción en la salida de la vejiga.
Parece que el RVU sin contaminación bacteriana y a baja presión no produce ningún daño a nivel renal, pero en presencia bacteriana representa un factor de riesgo para el desarrollo de pielonefritis.
Su incidencia se calcula entre el 0,4 y el 1,8% en niños aparentemente sanos y del 25 al 50% en los que han tenido un cuadro de pielonefritis46.
Tras sospecha prenatal, debe realizarse ECO a los 10 días para confirmar dilatación pieloureteral y, si es positiva, habrá que efectuar la CUMS para descartarlo47.
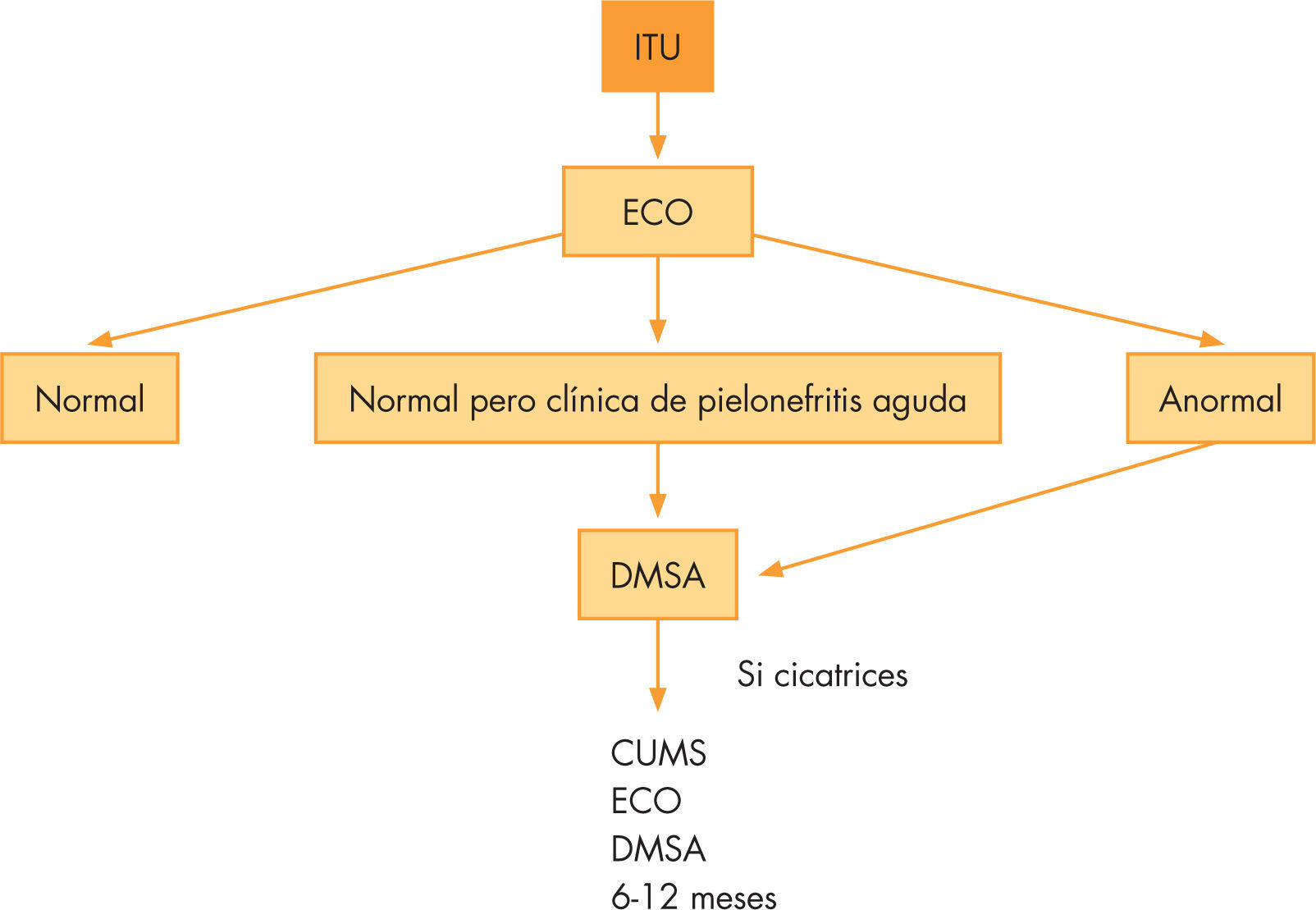
Las recomendaciones para estudios de imágenes en niños tras ITU por la Sociedad Europea de Radiología Pediátrica (2007) son las que se muestran en la figura 3 emsp48,49.

El objetivo del tratamiento es prevenir el desarrollo ITU recurrentes, pielonefritis y formación de cicatrices para prevenir la pérdida de función renal.
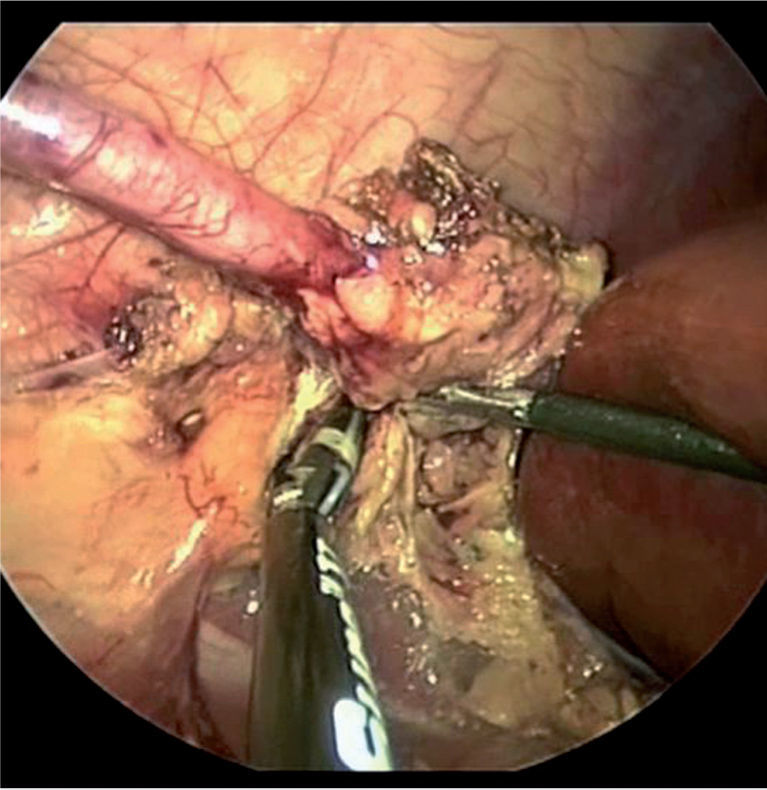
Válvulas de uretra posteriorLas VUP son la causa más frecuente de obstrucción congénita a la salida vesical en el niño, dando lugar a un amplio espectro de daños en todo el tracto urinario. Las válvulas son membranas obstructivas en la luz de la uretra, que se extienden desde el veru montanum distalmente (fig. 4). Solo ocurren en varones y son la primera causa de fallo renal y trasplante en la población pediátrica. La incidencia es de 1/5.000-8.000 varones. Últimamente, se ha referido en 1/1.250 en ecos fetales50.

Aunque el mecanismo productor de las VUP no es conocido se cree que se debe a una detención del desarrollo embriológico de la uretra entre las semanas 9 y 14, se han postulado diferentes teorías sobre la embriología de las VUP:
- –
Persistencia de la membrana urogenital con una anormal canalización de la uretra.
- –
- –
Anormal integración de los conductos de Wolf dentro de la uretra posterior, que se insertarían de manera anormal en la parte anterior de la cloaca, dando como resultado unos pliegues más gruesos, más prominentes y obstructivos.
Debido a que las VUP están presentes durante las primeras etapas del desarrollo fetal, los tejidos primitivos maduran en un medio ambiente anormal en el que hay alta presión intraluminal y distensión de los órganos. Como consecuencia, se provoca principalmente daño al parénquima renal y alteración del funcionamiento del músculo liso del uréter y de la vejiga. Estos cambios pueden persistir a pesar de que se alivie con éxito la obstrucción primaria y originar enfermedad renal crónica, ureterohidronefrosis, RVU y disfunción vesical.
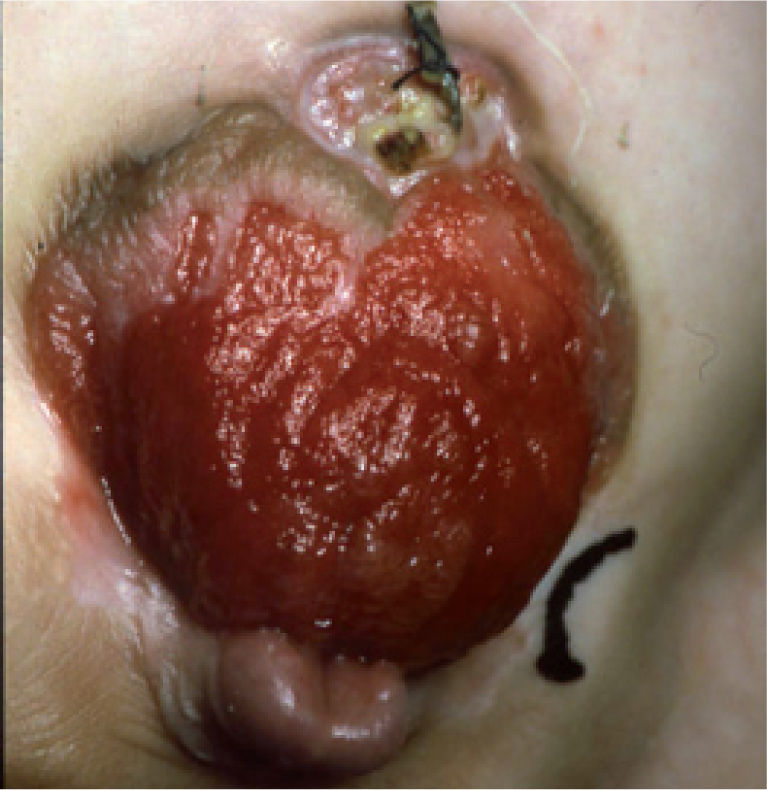
Extrofia vesicalLa extrofia vesical es una compleja anomalía congénita que engloba alteraciones en el sistema musculoesquelético, en el aparato genitourinario y en el tracto intestinal.
La extrofia de vejiga generalmente está incluida dentro del complejo extrofia-epispadias y está formada por un espectro de malformaciones embriológicas que incluye:
- –
Epispadias: la placa uretral está total o parcialmente abierta en la superficie dorsal del pene.
- –
Extrofia vesical clásica: la placa vesical está abierta en la parte inferior del abdomen y la uretra es epispádica (fig. 5).
- –
Extrofia de cloaca: la placa vesical y la porción ileocecal están unidas y abiertas, mostrándose como una placa única en la región inferior del abdomen.
- –
Entre las malformaciones anteriormente descritas existe un espectro de variantes que se sitúan entre ambas.

Los autores declaran que no existe conflicto de intereses.