








Puntos clave

Las inmunodeficiencias son un grupo de enfermedades muy amplio, con mecanismos patogénicos diferentes, que tienen en común un fallo en los mecanismos de regulación de la defensa inmunitaria frente a las agresiones exteriores. Este fallo en la regulación da lugar a enfermedades cuyo origen puede estar en la alteración de un receptor (TOLL), en la ausencia de una interleucina (IL), en la falta de función de un linfocito B o de un linfocito T, o en la falta de una proteína como la MBL (mannose-binding lectine). Al final, sea cual sea el mecanismo patogénico, la sintomatología predominante es la presencia de infecciones, aunque estos niños también van a presentar neoplasias de estirpe linfoide y enfermedades autoinmunes1,2.
Las inmunodeficiencias se clasifican en congénitas o primarias, casi siempre debidas a defectos de mutación o deleción de genes, que suelen ser hereditarias, pero también mutaciones de novo, y adquiridas o secundarias, debidas a una enfermedad que facilita la pérdida de anticuerpos o linfocitos, como la malnutrición, las diarreas crónicas o el síndrome nefrótico3.
En la actualidad, se han identificado más de 150 formas clínicas de inmunodeficiencias primarias. Gracias a los recientes avances en la biología molecular, se conocen los defectos moleculares responsables de una parte importante de las inmunodeficiencias primarias, con indudables ventajas en cuanto a la posibilidad de un diagnóstico de certeza, diagnóstico prenatal en las familias afectadas, consejo genético y posibilidad de tratamiento mediante terapia génica en algunos casos4,5.
Desarrollo de la inmunidad en el niñoEl desarrollo de la inmunidad en el niño se completa hacia los 10-12 años. Hasta entonces el sistema inmunitario, al igual que otros órganos y sistemas, es inmaduro y tras una serie de estímulos y respuestas adaptativas al ambiente exterior, llega a desarrollarse totalmente, con capacidad de regulación y respuesta frente a agentes externos extraños (microorganismos), de mantenimiento de la homeostasias y reconocimiento del propio individuo.
Los componentes del sistema inmunitario, timo, ganglios linfáticos, bazo, células linfoides y anticuerpos, están presentes en el feto en períodos tempranos. Desde la sexta semana de gestación se reconoce la formación del timo y a los 38 días de la concepción se puede detectar IgG materna. Potencialmente existe capacidad de responder, por parte del sistema inmunitario, frente a estimulaciones antigénicas. Pero el feto está en un ambiente estéril que no precisa una respuesta inmunitaria puntual. Al nacer tras los estímulos antigénicos adecuados, el sistema inmunitario entrará en una gran actividad, que poco a poco irá desarrollándose por completo, hasta igualarse a la del adulto6.


La primera línea defensiva ante una agresión exterior, constituye la «inmunidad de barrera», integrada por la piel, las mucosas, la barrera hematoencefálica y los movimientos mecánicos de expulsión de sustancias nocivas o de desecho, al exterior.
La inmunidad de barrera en el recién nacido, y más en el prematuro, es deficiente.
La piel es fina, fácilmente friable, tiene poca capacidad para inflamarse, no tienen secreción grasa y las glándulas de los folículos pilosos no son funcionantes. No se produce sudor y esto confiere a la piel un pH alcalino, en el que las bacterias colonizan con facilidad. La defensa de barrera mucosa se encuentra en el aparato respiratorio y el aparato digestivo, puerta de entrada habitual de gérmenes. El recién nacido tiene poca capacidad para toser, secreta poco moco, los movimientos ciliares son escasos y se afectan muy precozmente por irritantes exteriores, como el humo del tabaco. La capacidad de formar anticuerpos IgA secretores es muy deficiente o nula en el período neonatal.
En el aparato digestivo la defensa de barrera está constituida por los movimientos peristálticos, el pH ácido gástrico, que impide la colonización bacteriana y la contaminación intestinal por coli no patógenos, que inhibe por competencia la colonización de otras bacterias patógenas. Mecanismos más específicos los forman los anticuerpos secretores de clase IgM, IgA o IgG producidos in situ por los linfocitos de las placas de Peyer.
La inmunidad inespecífica neonatal es defectuosa. Esta situación hace que en el período neonatal se den con facilidad infecciones por gérmenes gramnegativos, estreptococo B, estafilococo y Pseudomonas. También por agentes infectantes intracelulares, como Toxoplasma. Y que exista deficiente respuesta a antígenos de polisacáridos neumocócicos o de Haemophilus influenzae.
Lectura rápida
La principal manifestación de una inmunodeficiencia es la susceptibilidad aumentada a las infecciones. Están producidas por gérmenes habituales o por gérmenes oportunistas de baja virulencia. Los signos de alarma van a ser diferentes según el tipo de defecto inmunológico, con una edad de inicio de los síntomas, unas manifestaciones y unos síntomas asociados particulares para cada forma de IDP. Los síntomas y signos clínicos de sospecha de inmunodeficiencia varían en función de la edad.
— Inmunidad específica humoral: está constituida por anticuerpos específicos, que forman las inmunoglobulinas. Al nacimiento el neonato tiene un 10% más de IgG que su madre y a las 3 semanas de vida, sintetiza IgA secretora. Pero existe deficiente formación de anticuerpos frente a antígenos polisacáridos, sobre todo los de clase IgG2, lo que facilita en este período de la vida las infecciones por estreptococo B. Ante cualquier infección el recién nacido y el feto responden con formación de anticuerpos IgM, que son de producción rápida, pero de corta vida media.
Existe una situación de riesgo infeccioso, aumentada, en los prematuros de menos de 30 semanas, en los que la IgG materna no ha tenido tiempo de atravesar la placenta, sobre todo si la tasa de IgG total es inferior a 400mg/dl. Los recién nacidos a término hijos de madre con hipo o agammaglobulinemia están en la misma situación. También los recién nacidos de bajo peso para su edad gestacional, por disfunción placentaria, que afecta al paso correcto a su través de IgG materna. Los recién nacidos con infección prenatal, viral, bacteriana o parasitaria tendrán una tasa de IgM al nacer elevada, siempre superior a 40mg/dl, si la infección se produce en el período en que el sistema inmunitario fetal tiene capacidad de responder.
El paso placentario de anticuerpos maternos de clase IgG no es uniforme para todos ellos. Anticuerpos protectores frente a determinadas infecciones presentadas por la madre tienen un paso elevado.
— Inmunidad específica celular: en el recién nacido, los linfocitos T circulantes están aumentados en número, con relación al adulto, también lo están los linfocitos B y los nulos, pero son inmaduros y expresan antígenos de superficie CD38 y CD45 que demuestran su inmadurez. La producción de linfocinas está descendida, sobre todo de interleucinas (IL) IL-3, IL-4 e IL-5 y está muy descendida la producción de interferón gamma. La función citotóxica está disminuida entre un 30 y un 60% con relación al adulto. Determinadas circunstancias en la vida del recién nacido pueden deprimir más la precaria inmunidad celular específica del neonato, que es de producción propia. Así, las infecciones virales, la hiperbilirrubinemia o el uso de fármacos inmunosupresores, en el propio niño, o que hayan sido administrados a su madre.
Lectura rápida
Retraso pondoestatural; diarrea crónica resistente y/o malabsorción; muguet persistente; infecciones por gérmenes oportunistas; neumonía antes de los 3 meses de vida; infecciones que no se resuelven con antibióticos intravenosos; tratamiento con antibióticos durante 2 meses o más con poco efecto; historia familiar de inmunodeficiencia primaria; reacciones sistémicas ante vacunas de gérmenes vivos; ausencia de sombra tímica en la radiografía de tórax; hipoplasia de tejido linfoide (amígdalas, ganglios).
Por la edad de presentación se debe sospechar como primera opción una inmunodeficiencia celular o combinada grave. En esta situación, las pruebas complementarias a practicar y los hallazgos que confirmarán el diagnóstico serán: en el hemograma: linfocitopenia, que puede asociar eosinofilia; poblaciones linfoides: linfocitopenia T (< 1.000/μl) con descenso tanto de CD4 como de CD8, linfocitos B y NK normales o disminuidos; hipogammaglobulinemia (IgA, IgG, IgM) de intensidad variable (pueden persistir las IgG maternas durante los primeros 3-6 meses de vida).
A partir del nacimiento y con los estímulos antigénicos constantes, producidos por las infecciones, las inmunizaciones del calendario vacunal y el contacto con proteínas y agentes ambientales nuevos, el sistema inmunitario inicia una serie de respuestas adaptativas que culminarán hacia los 10-12 años, con la maduración de la inmunidad celular, humoral e inespecífica, que a esta edad es similar a la del adulto.
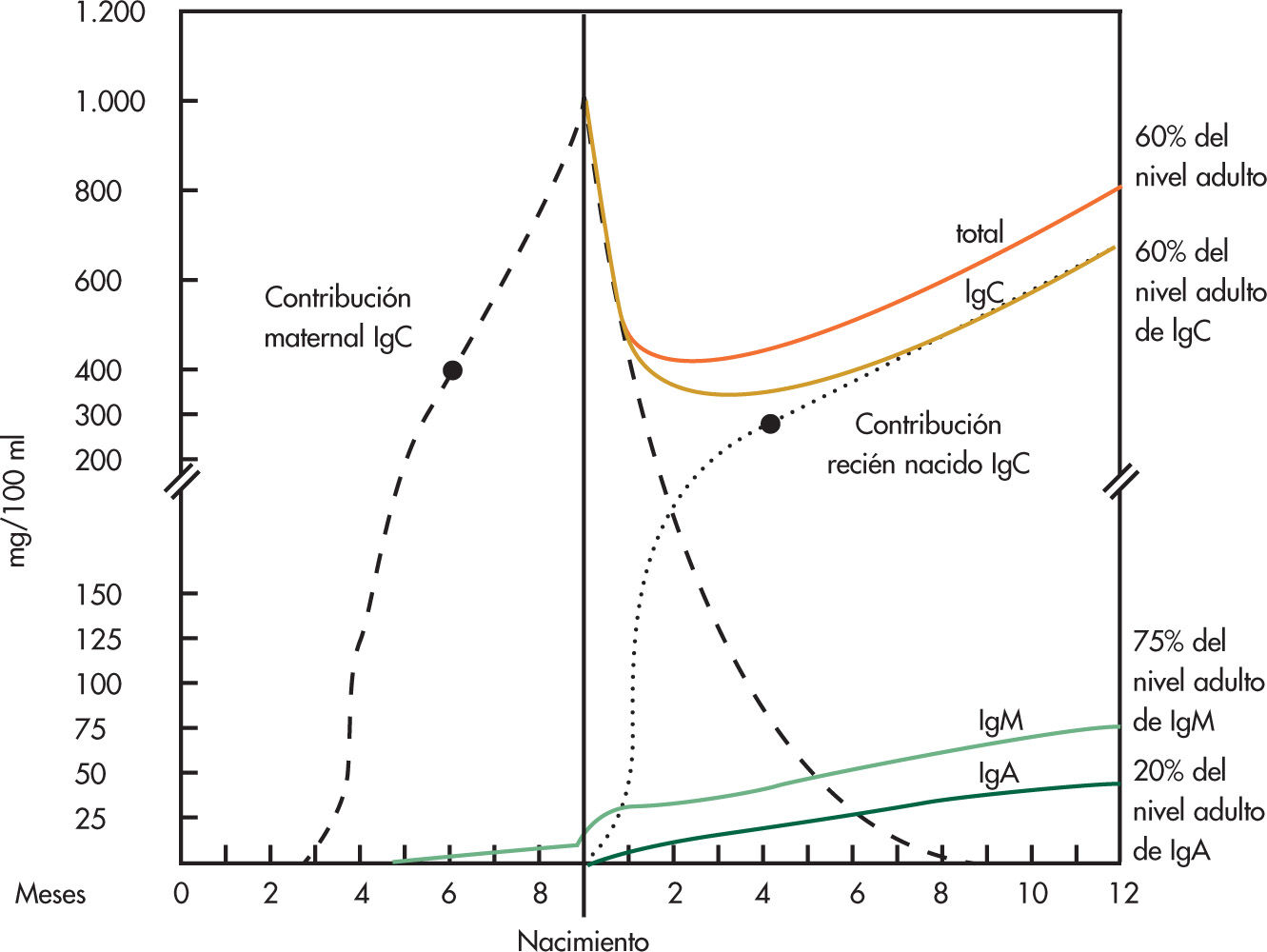
Los mayores cambios se producen durante el primer año de vida y son perceptibles sobre todo en la inmunidad humoral específica, mediada por anticuerpos (inmunoglobulinas). Después del nacimiento, se inicia de forma rápida la destrucción de la IgG materna que pasó a través de la placenta, de tal forma que hacia el tercer mes de vida el lactante tiene unos 300mg/dl de IgG materna, de los 1.000mg/dl que tenía al nacer. La IgG propia del niño se produce también de forma rápida, pero a los 3 meses de vida tiene una tasa baja, entre 200 y 300mg/dl. Este período de la vida, entre los 2 y 4 meses, es especialmente susceptible a las infecciones. La IgG materna desaparece totalmente hacia los 9 meses de vida y a los 12 meses el lactante tiene una IgG propia total, de unos 700mg/dl, el 60% de la del adulto. La IgM es la inmunoglobulina propia del niño, ya que no atraviesa la placenta, al nacer es de unos 40mg/dl, y en la vida posnatal experimenta un crecimiento rápido y progresivo de forma que a los 12 meses el niño tiene ya el 75-80% de la del adulto, aproximadamente 60-70mg/ dl. Los lactantes sanos que reciben estímulos antigénicos intensos y repetidos, como los que asisten a guarderías, tienen una producción más rápida e intensa de IgM, como expresión de una buena respuesta de su sistema inmunitario.
La IgA es la inmunoglobulina más especializada, es propia del lactante ya que no atraviesa la placenta y su producción se inicia en la vida posnatal de forma lenta y progresiva, de tal manera que a los 12 meses el niño tiene sólo el 20% de la tasa del adulto, entre 25-50mg/dl. La maduración de la IgA se completa hacia los 10-12 años (fig. 1).

La exploración de la inmunidad en el niño puede resultar muy compleja según la sospecha diagnóstica. Exponemos una sencilla pauta, que puede ayudar al diagnóstico definitivo de una inmunodeficiencia, a la demostración de una inmunidad normal o a la orientación hacia inmunodeficiencias parciales o raras, en las que es preciso realizar pruebas especiales como biopsias, estudio genético, determinación de receptores de células o IL.
Formas clínicas de inmunodeficiencias primariasLas IDP se clasifican según el área del sistema inmunitario afectada. Las 4 formas principales (tabla 1) son:
- 1.
Defecto del linfocito B: inmunodeficiencia humoral o déficit de anticuerpos, que constituye el 70% de todas las IDP7,8.
- 2.
Defecto del linfocito T: inmunodeficiencia celular. También recibe el nombre de inmunodeficiencia combinada porque el linfocito T colabora en la activación del linfocito B para la generación de anticuerpos, por lo que en esta forma de inmunodeficiencia quedan afectadas ambas respuestas, la celular primariamente y la humoral secundariamente. Es la forma más grave de IDP. Constituye el 15% de todas las IDP9–11.
- 3.
Defecto de los fagocitos, en su función y/o número, que constituye el 5% de todas las IDP12.
- 4.
Defecto del complemento, que constituye el 10% de todas las IDP.
Características principales de las inmunodeficiencias primarias
| Forma de IDP | Ejemplos de enfermedades | Edad de inicio | Complicaciones infecciosas | Otras manifestaciones |
|---|---|---|---|---|
| Inmunodeficiencia predominante de anticuerpos (humoral) |
| > 6 meses de vida. Puede manifestarse en la adolescencia o en la edad adulta |
|
|
| Inmunodeficiencias combinadas (humorales y celulares) |
| < 6 meses de vida |
|
|
| Defectos de los fagocitos |
| Lactante o infancia |
|
|
| Defectos del complemento |
| Cualquier edad |
|
|
AR: autosómica recesiva; CMV: citomegalovirus; ID: inmunodeficiencia; IDCG: inmunodeficiencia combinada grave; IDP: inmunodeficiencia primaria; Ig: inmunoglobulina; VEB: virus de Epstein-Barr.
Existe un grupo de IDP, que forman parte de síndromes bien conocidos, con una asociación de síntomas y signos que permiten la identificación clínica con cierta facilidad. En ocasiones la inmunodeficiencia es el primer signo, acompañado de infecciones, pero en otras ocasiones será una malformación congénita, como una cardiopatía conotruncal, o una ataxia progresiva, lo que orienta el diagnóstico. Es importante reconocer estas formas de inmunodeficiencia asociadas a defectos mayores, para el tratamiento precoz y evitar el deterioro de la salud del niño, así como realizar el estudio genético que en la mayoría de estos síndromes es conocido (tabla 2)13.
Síndromes asociados a inmunodeficiencias primarias
| Síndrome | Manifestaciones clínicas indicativas del síndrome | Déficit inmunológico asociado | Alteraciones de laboratorio |
|---|---|---|---|
| Wiskott-Aldrich |
|
|
|
| Ataxiatelangiectasia |
|
|
|
| Sindrome de Di George |
|
|
|
| Chediak-Higashi y Griscelli |
|
|
|
Ag: antígenos; AR: autosómica recesiva; CD3 +: linfocitos T totales; CD4+: linfocitos T4; ID: inmunodeficiencia; Ig: inmunoglobulinas; IgA: inmunoglobulina A; IgM: inmunoglobulina M; NK: células naturales citotóxicas; T: linfocitos T.
El diagnóstico precoz de las IDP es fundamental para poder instaurar un tratamiento eficaz que evite la aparición de daño irreversible en diferentes órganos, responsable de la morbilidad y mortalidad de los pacientes afectados. Es labor del pediatra general la sospecha y la orientación inicial, con pruebas de laboratorio sencillas y de precio asequible, de un caso de IDP para luego derivar el paciente al especialista, que completará el diagnóstico inmunológico y genético e instaurará un tratamiento específico.
Las IDP fueron originalmente catalogadas como enfermedades raras con una expresión clínica grave temprana en la vida del niño. Las manifestaciones clínicas pueden ser en ocasiones escasas y no exclusivamente infecciosas, y pueden manifestarse en la adolescencia o la edad adulta como ocurre en la inmunodeficiencia variable común14–16.
La sospecha de una IDP empieza con una correcta historia clínica con especial hincapié en los antecedentes familiares de consanguinidad, muerte prematura no explicada o déficit inmunológico, dado que la gran mayoría de las IDP son enfermedades hereditarias, principalmente autosómicas recesivas o ligadas al X.
La principal manifestación de una inmunodeficiencia es la susceptibilidad aumentada a las infecciones, que pueden ser frecuentes, duraderas, graves y con tendencia a complicarse, y estar producidas por gérmenes habituales u oportunistas de baja virulencia. Los signos de alarma van a ser diferentes según el tipo de defecto inmunológico, con una edad de inicio de la clínica, unas manifestaciones y unos síntomas asociados particulares para cada forma de IDP (v. tabla 1). En el 87% de los casos van a ser las complicaciones infecciosas las que van a dar el signo de alarma que llevará al diagnóstico de IDP17.
Ante la sospecha de una IDP, existen pruebas de laboratorio accesibles que el pediatra general puede solicitar. Dichas pruebas pueden ser fuente de valiosa información para un primer diagnóstico sindrómico. En un segundo tiempo, y ya con la sospecha de una de las 4 formas de IDP, los análisis pueden ser más extensos aun siendo exámenes de primera línea. Dichos exámenes y las alteraciones características en cada forma de IDP se irán exponiendo a continuación18.
Para la mejor orientación clínica de una IDP se describen a continuación los síntomas y signos clínicos de sospecha en función de la edad del niño, y a continuación la propuesta de pruebas complementarias a practicar para la confirmación diagnóstica.
Lectura rápida
Ocho o más episodios de otitis media en un año, sobre todo si la otitis se cronifica, los episodios repetidos persisten por encima de los 5 años, o si se asocian a infecciones broncopulmonares y/o sinusitis; 2 o más episodios de sinusitis grave en un año; 2 o más episodios de neumonía en un año; 2 o más infecciones invasivas como meningitis, celulitis, mastoiditis, osteomielitis o sepsis; infecciones que no se resuelven con antibióticos intravenosos; tratamiento con antibióticos durante 2 meses o más con poco efecto; enfermedades alérgicas, autoinmunes o síndromes asociados a inmunodeficiencia; neoplasias de origen linfático; historia familiar de inmunodeficiencia primaria; reacciones sistémicas ante vacunas de gérmenes vivos. En esta situación, las pruebas complementarias a practicar y los hallazgos que confirmarán el diagnóstico serán: hemograma normal o mínima linfocitopenia; hipogammaglobulinemia (global o de un subtipo concreto de inmunoglobulinas); poblaciones linfoides: linfocitos B normales o disminuidos; medición de la formación de anticuerpos (proteicos y polisacáridos).
Son datos de sospecha de inmunodeficiencia los siguientes19:
- —
Retraso pondoestatural.
- —
Diarrea crónica resistente y/o malabsorción.
- —
Muguet persistente.
- —
Infecciones por gérmenes oportunistas.
- —
Neumonía antes de los 3 meses de vida.
- —
Infecciones que no se resuelven con antibióticos intravenosos.
- —
Tratamiento con antibióticos durante 2 meses o más con poco efecto.
- —
Historia familiar de IDP.
- —
Reacciones sistémicas ante vacunas de gérmenes vivos.
- —
Ausencia de sombra tímica en la radiografía de tórax.
- —
Hipoplasia de tejido linfoide (amígdalas, ganglios).
Lectura rápida
La presencia de 2 o más infecciones invasivas como meningitis o sepsis por Neisseria hará pensar en un defecto en el complemento (C5-C9), para lo que se debe solicitar la determinación de CH50, que es el 50% del complemento hemolítico total. La presencia de: abscesos recurrentes cutáneos, pulmonares o en órganos profundos (bazo, hígado); caída retardada del cordón umbilical; dificultad en la cicatrización; periodontitis/estomatitis persistente; fiebre mantenida; infecciones que no se resuelven con antibióticos intravenosos; historia familiar de inmunodeficiencia primaria.
Por la edad de presentación se debe sospechar como primera opción una inmunodeficiencia celular o combinada grave. En esta situación, las pruebas complementarias que se deben practicar y los hallazgos que confirmarán el diagnóstico son:
- —
Hemograma: linfocitopenia. Puede asociar eosinofilia (síndrome de Omenn). Normalmente en los niños las células T constituyen el 55-80% de linfocitos periféricos, y el número de CD4 es 1,5-2 veces superior a los CD8. Dado que la mayoría de linfocitos circulantes son T, en la inmunodeficiencia combinada grave (que cursa con una linfocitopenia T), se apreciará claramente una linfocitopenia en el hemograma. En niños de 6 meses aproximadamente una linfocitopenia total < 1.000/μl debe obligar a descartar una inmunodeficiencia combinada grave. Aunque las inmunoglobulinas pueden ser inicialmente normales (IgG materna) no hay formación de anticuerpos después de la vacunación.
- —
Poblaciones linfoides: linfocitopenia T (< 1.000/μl) con descenso tanto de CD4 como de CD8. Linfocitos B y NK normales o disminuidos.
- —
Hipogammaglobulinemia (IgA, IgG, IgM) de intensidad variable: pueden persistir las IgG maternas durante los primeros 3-6 meses de vida.
Se debe valorar como signo sospechoso de inmunodeficiencia la presencia de:
- —
Ocho o más episodios de otitis media en un año, sobre todo si la otitis se cronifica, los episodios repetidos persisten por encima de los 5 años, o si se asocian a infecciones broncopulmonares y/o sinusitis.
- —
Dos o más episodios de sinusitis grave en un año.
- —
Dos o más episodios de neumonía en un año.
- —
Dos o más infecciones invasivas como meningitis, celulitis, mastoiditis, osteomielitis o sepsis.
- —
Infecciones que no se resuelven con antibióticos intravenosos.
- —
Tratamiento con antibióticos durante 2 meses o más con poco efecto.
- —
Enfermedades alérgicas, autoinmunes o síndromes asociados a inmunodeficiencia.
- —
Neoplasias de origen linfático20.
- —
Historia familiar de inmunodeficiencia primaria.
- —
Reacciones sistémicas ante vacunas de gérmenes vivos.
Por la edad de presentación, la sospecha diagnóstica en primera opción será una inmunodeficiencia humoral específica por deficiencia de anticuerpos (inmunodeficiencia transitoria, agammaglobulinemia e inmunodeficiencia variable común son las más frecuentes). Las pruebas complementarias que se deben practicar y los hallazgos que confirmarán el diagnóstico son:
- —
Hemograma: normal o mínima linfocitopenia.
- —
Hipogammaglobulinemia (global o de un subtipo concreto de inmunoglobulinas): cuando se encuentra un nivel bajo de IgG en el lactante, con frecuencia se trata de una hipogammaglobulinemia transitoria asociada a un defecto en la maduración del sistema inmunitario. En la hipogammaglobulinemia transitoria de la infancia los valores de IgG son bajos (200-400mg/ dl), y este hecho puede prolongarse hasta los 3-4 años. Las cifras de IgM e IgA suelen estar disminuidas o normales. Pero un nivel bajo de IgG puede ser el primer signo de una inmunodeficiencia grave como la agammaglobulinemia de Bruton, en la que existe un descenso profundo de todas las clases de inmunoglobulinas. Para diferenciar estas 2 entidades es fundamental medir la formación de anticuerpos. En los escolares y adolescentes, en cambio, un déficit de IgG es indicativo de una inmunodeficiencia variable común. En la inmunodeficiencia variable común las cifras de IgG son bajas (siempre menores de 500mg/dl); por lo general, los niveles de IgM están conservados. Se puede asociar a un defecto en la síntesis de anticuerpos específicos, sobre todo frente a los polisacáridos.
Un déficit de inmunoglobulinas también puede ser debido a pérdidas proteicas, por lo que se recomienda medir la albúmina sérica.
- —
Poblaciones linfoides: linfocitos B normales o disminuidos. En la agammaglobulinemia de Bruton existe sólo un 1-2% de linfocitos B circulantes, a diferencia de la inmunodeficiencia variable común en la que la cifra de linfocitos B circulantes es normal o ligeramente descendida.
- —
Medición de la formación de anticuerpos (proteicos y polisacáridos): en las deficiencias de inmunoglobulinas se debe estudiar la funcionalidad de la respuesta humoral. La medición de la formación de anticuerpos en respuesta al antígeno puede ser de 2 tipos: respuesta frente a antígenos proteicos y frente a antígenos polisacáridos. Los mecanismos inmunológicos para generar cada una de las 2 respuestas son diferentes (T-dependiente en antígenos proteicos y T-independiente en antígenos polisacáridos), por ello deberán evaluarse ambas, teniendo en cuenta que la respuesta a antígenos polisacáridos aparece en condiciones normales por encima de los 18-24 meses de vida. Ejemplos de antígenos polisacáridos son: la vacuna neumocócica no conjugada o 23-valente o los antígenos del grupo sanguíneo ABO. Por ello una forma de medir la respuesta frente a antígenos polisacáridos es mediante la cuantificación de títulos de isohemaglutininas o anticuerpos anti-ABO. Pacientes con grupo sanguíneo del tipo A poseen anti-B, los B anti-A y los O ambos, los AB no forman isohemaglutininas. Se deben encontrar valores mayores de 1:10. Son de clase IgM.
En general, la forma más sencilla de evaluar la formación de anticuerpos en la edad pediátrica es mediante el estudio de la respuesta frente a las vacunas del calendario vacunal. La formación correcta de anticuerpos vacunales frente a antígenos proteicos permite excluir un defecto inmunológico significativo dado que ello requiere de la presencia de linfocitos B, de inmunoglobulinas y de una respuesta de linfocitos T CD4+. Ejemplos de antígenos proteicos son el toxoide tetánico y la difteria.
Lectura rápida
La sospecha diagnóstica será de una neutropenia o de un defecto en la fagocitosis y las pruebas diagnósticas que se deben practicar son: hemograma: neutropenia/neutrofilia; estudio de inmunoglobulinas: normal.
En los niños con infecciones bacterianas oportunistas de repetición e infecciones fúngicas, pero con neutrófilos normales o altos e inmunoglobulinas normales o aumentadas, se debe pensar en enfermedad granulomatosa crónica. Si estos pacientes presentan neutrófilos altos de forma persistente, y caída retrasada del cordón umbilical, se debe pensar en déficit de adhesión leucocitaria.

A cualquier edad la presencia de 2 o más infecciones invasivas como meningitis o sepsis por Neisseria debe hacer pensar en un defecto en el complemento (C5-C9), para lo que se debe solicitar la determinación de CH50, que es el 50% del complemento hemolítico total19.
La actividad del complemento es muy termolábil, por lo tanto las muestras deben ser transportadas en hielo.
El CH50 mide la vía clásica del complemento. Por ello los defectos de properdina a factor D, que pertenecen a la vía alternativa, deben detectarse con la medición de la vía alternativa (APH50). Si el CH50 es nulo, puede tratarse de un déficit de C1 a C8. En este caso, se deberán determinar los niveles de C3 y C4. Niveles de C3 y C4 normales con CH50 indetectable ponen de manifiesto déficit de complemento congénito; entonces se debe determinar el resto de componentes del complemento. Niveles de CH50 y C3 y/o C4 bajos deben hacer pensar en un consumo de complemento (un nivel bajo de complemento puede ser secundario a una infección).
En el déficit de C9, el valor de CH50 no es nulo, sino del 25 al 50%.
A cualquier edad, la presencia de:
- —
Abscesos recurrentes cutáneos, pulmonares o en órganos profundos (bazo, hígado).
- —
Caída retardada del cordón umbilical.
- —
Dificultad en la cicatrización.
- —
Periodontitis/estomatitis persistente.
- —
Fiebre mantenida.
- —
Infecciones que no se resuelven con antibióticos intravenosos.
- —
Historia familiar de inmunodeficiencia primaria21–23.
La sospecha diagnóstica será de una neutropenia o de un defecto en la fagocitosis. Las pruebas diagnósticas son:
- —
Hemograma: neutropenia/neutrofilia.
- —
Estudio de inmunoglobulinas: normal (excepto en síndrome de hiper-IgE o de Job).
En los niños con infecciones bacterianas oportunistas de repetición e infecciones fúngicas, pero con neutrófilos normales o altos e inmunoglobulinas normales o aumentadas, se debe pensar en enfermedad granulomatosa crónica (defecto de oxidación intracelular de los neutrófilos) cuyo diagnóstico se confirmará con un test de quimioluminiscencia.
Si estos pacientes presentan neutrófilos altos de forma persistente, y caída retrasada del cordón umbilical, se debe pensar en déficit de adhesión leucocitaria, cuyo diagnóstico se confirmará con el estudio de las glucoproteínas de superficie de neutrófilos (CD11b/ CD18 y CD15).