








De forma genérica, los síndromes clínicos que resultan de las alteraciones de la hemoglobina (Hb) se conocen como hemoglobinopatías y para conocerlos se interpretan diversas pruebas de laboratorio, de las cuales la más relevante es la electroforesis (EF) de Hb.
La molécula de Hb humana1 está formada por 2 pares de cadenas de globina, y a cada una de ellas se une una molécula de hemo. Fisiológicamente, se forman 6 variantes:
- –
Tres variantes transitorias embrionarias, denominadas Hb Gower 1, Hb Gower 2 y Hb Portland.
- –
HbF, que es la que predomina en la vida fetal.
- –
HbA, que constituye más del 95% de la Hb del adulto.
- –
HbA2, también presente en un porcentaje menor (1–3,5%) en el niño y en el adulto.
Puntos clave

Las cadenas de globina que forman la molécula de Hb posnatalmente se asocian con la forma de un tetrámero, y se designan como α, β, χ y δ. Así, las variantes de Hb se forman por las siguientes cadenas hasta un total de 4:
- –
HbA: α2β2.
- –
HbF: β2χ2.
- –
HbA2: α2δ2.
Hay muchas variantes de Hb determinadas genéticamente (heredadas)2 y aunque la mayoría son inocuas, otras ocasionan alteraciones clínicas notables. Las hemoglobinopatías pueden agruparse en 3 categorías fundamentales, las más relevantes son las 2 primeras:
- –
Variantes estructurales de la Hb, como la HbS (anemia falciforme o drepanocitosis). Se atribuyen a la sustitución de aminoácidos en las cadenas de globina. Se han descrito casi un millar, pero la mayoría son un hallazgo casual y clínicamente silentes. Las que ocasionan enfermedad se deben a la sustitución de aminoácidos que conlleva cambios en la estructura secundaria o terciaria del tetrámero de Hb.
- –
Fallo en la síntesis de una cantidad adecuada de Hb normal (talasemias) por defecto en la producción de 1 o más de las cadenas de globina. La clínica asociada surge de la combinación de una inadecuada síntesis de Hb y de la acumulación de cadenas de globina que no han podido formar el tetrámero. Lo primero causa anemia y microcitosis, y lo segundo, eritropoyesis ineficaz y hemólisis.
- –
Fallo en el salto normal que se produce en el neonato de producir HbF a HbA (persistencia hereditaria de Hb fetal).
La investigación de los pacientes con sospecha de sufrir una hemoglobinopatía debe incluir una anamnesis completa con antecedentes familiares, exploración física y análisis de laboratorio. Estas pruebas se definieron ya en 1975 por un panel de expertos del Comité Internacional para la Estandarización en Hematología3–5: contaje de serie roja con índices; Hb; hematocrito; frotis de sangre periférica; reticulocitos; metabolismo del hierro; test de solubilidad; test de falciformación; cuantificación de HbA2 y HbF, y EF de Hb a pH alcalino. Si la Hb detectada es inestable o con afinidad por el oxígeno alterada, hay que añadir el test de estabilidad térmica (por calor) y química (isopropanol). De este modo, se puede hacer un diagnóstico fiable en la mayoría de los casos, aunque a veces hay que recurrir a técnicas más sofisticadas como la EF a pH ácido, el estudio de globinas, el isoelectroenfoque o incluso el análisis molecular de ADN.
Aunque las EF son muy utilizadas, en la actualidad muchas veces están desplazadas por la cromatografía líquida de alta resolución (HPLC)6, que es muy útil para cuantificar de forma rápida HbA2 y HbF, así como para identificar variantes estructurales. Este método es imprescindible cuando se trata de analizar gran cantidad de muestras en poco tiempo, como en el caso del cribado neonatal de anemia falciforme.
Electroforesis en acetato de celulosa a pH alcalino1,7Rutinariamente se realiza este tipo de EF a pH 8,4-8,6 usando una membrana de acetato de celulosa como sustrato, y es un método simple, rápido y sensible. Detecta las variantes de Hb más comunes, y tradicionalmente es la técnica más utilizada para la evaluación inicial.
A pH alcalino, la Hb es una proteína cargada negativamente y que en un campo eléctrico migra hacia el ánodo (+). Las variantes de Hb con distintas cargas en su superficie se separan de la HbA y, por tanto, todas las detectadas son diferentes de ella. Pueden haber Hb anormales sin cambio en la carga, y por tanto no serán diferenciadas
La figura 1 muestra las variantes de Hb más frecuentes detectadas por esta técnica y puede observarse que se obtiene una separación de las HbC, S, F, A y J. Cuando se detecta una variante es útil cuantificar el porcentaje estimado por un procedimiento similar al explicado más adelante para la HbA2 o por HPLC.

- –
Puede ser difícil detectar bandas menores como Hb Constant Spring, HbA2, Hb Bart y HbH.
- –
En el neonato, en el que hay pequeñas cantidades de HbA y enormes de HbF, puede ser difícil detectar HbA. Por eso siempre hay que recurrir a la EF en agar citrato cuando no se detecte HbA, o utilizar isoelectroenfoque.
- –
No es posible diferenciar entre HbS, D y G. No es posible diferenciar entre HbC, E y O. Por ello, todas las muestras en las que se detecta una sola banda en la posición de la HbS o C también se deben analizar en agar citrato o con isoelectroenfoque para excluir la presencia de heterocigotos compuestos como HbSD, SG, CE o CO.
Es muy efectiva en la detección inicial de variantes de Hb en combinación con la anterior. La mayoría de las Hb tiende a desplazarse igual que la HbA, y las que lo hacen de forma distinta es porque el aminoácido sustituido está relacionado con la unión al 2,3 DPG. Los factores que intervienen en la separación de las Hb son, además de la carga eléctrica, las interacciones fisicoquímicas entre la fase sólida (agar) y las moléculas de Hb, que dependen de la localización superficial de la mutación.
La EF en agar citrato se realiza a pH 6 y presenta más problemas técnicos que la anterior. Diferencia a la HbC, S, A y F, y en ella corren igual que la A las HbD, E, G, lepore, H y J; la Bart's se sitúa en la misma posición que la F. En acetato de celulosa a pH 6,5 se separan las HbH y Bart's de otras variantes de movilidad rápida.
Estimación de la HbA2El aumento en la HbA2 es característico de la β talasemia heterocigota. Se pueden hacer estimaciones tras elución desde una EF en acetato de celulosa o por cromatografía, ya sea en microcolumna o por HPLC9,10.
- –
Elución desde acetato de celulosa: la separación entre HbS y A2 cuando la primera está presente, está disminuida. Igualmente, este método no es apropiado en presencia de HbC, E y O, ya que la movilidad de éstas es igual que la de HbA2.
- –
Cromatografía en microcolumna: depende de la interacción de los grupos con carga de la resina de intercambio iónico de la columna con los grupos de la molécula de Hb. La separación de los componentes depende del pH, la carga iónica del buffer que se inyecta en la columna, el tipo de resina, el volumen que se añade y el flujo.
- –
HPLC: el mecanismo es similar al anterior y las fracciones eludías se detectan por un detector de luz ultravioleta/visible que integra los resultados a través de un ordenador. El procedimiento es muy rápido, exacto y reproducible.
- –
Mayores del 8%: estos valores son raros. Repetir y excluir una variante estructural.
- –
Entre el 3,8 y el 8%: rasgo β-talasemia, Hb inestables.
- –
Entre el 3,4 y el 3,7%: rasgo β-talasemia con ferropenia, rasgo β-talasemia con variante adicional de variante de cadena δ, interacción de α y β-talasemia, difícil medición por presencia de HbS, interacción de α-talasemia y HbS, error analítico.
- –
Menores del 3,5%: resultado normal, δβ talasemia, raros casos de rasgo β-talasemia, α talasemia, enfermedad de la HbH.
La HbF puede estimarse por diversos métodos basados en su resistencia a la desnaturalización a pH alcalino o por HPLC.
Interpretación de los valores de HbF- –
Entre el 0,2 y el 1%: normal.
- –
Entre el 1 y el 5%: en 30-50% de los rasgos β-talasemia, algunas variantes heterocigotas y homocigotas de Hb, algunos heterocigotos compuestos de variantes de Hb y rasgo β-talasemia, algunas enfermedades hematológicas (aplasias, mielodisplasias, etc.), esporádico en algunas poblaciones afrocaribeñas.
- –
Entre el 5 y el 20%: ocasionalmente en rasgo β-talasemia, algunas variantes de Hb homocigotas, algunos heterocigotos compuestos de una variante de Hb y rasgo β talasemia, algunos tipos de persistencia hereditaria de HbF heterocigotos, δβ talasemia.
- –
Entre el 15 y el 45%: algunos casos de β talasemia intermedia, persistencia heterocigota de HbF tipo africano.
- –
Mayores del 45%: β talasemia major, algunos casos de intermedia, neonatos, persistencia homocigota de HbF africana.
Tras la completa historia clínica se aconseja seguir el esquema de la figura 2 y tablas 1 y 2.

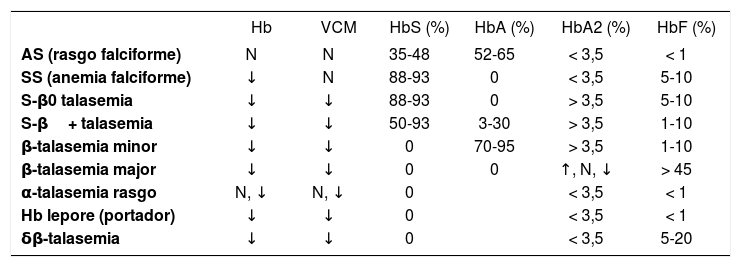
Resultados en el hemograma y electroforesis en variantes de HbS y talasemia
| Hb | VCM | HbS (%) | HbA (%) | HbA2 (%) | HbF (%) | |
|---|---|---|---|---|---|---|
| AS (rasgo falciforme) | N | N | 35-48 | 52-65 | < 3,5 | < 1 |
| SS (anemia falciforme) | ↓ | N | 88-93 | 0 | < 3,5 | 5-10 |
| S-β0 talasemia | ↓ | ↓ | 88-93 | 0 | > 3,5 | 5-10 |
| S-β+ talasemia | ↓ | ↓ | 50-93 | 3-30 | > 3,5 | 1-10 |
| β-talasemia minor | ↓ | ↓ | 0 | 70-95 | > 3,5 | 1-10 |
| β-talasemia major | ↓ | ↓ | 0 | 0 | ↑, N, ↓ | > 45 |
| α-talasemia rasgo | N, ↓ | N, ↓ | 0 | < 3,5 | < 1 | |
| Hb lepore (portador) | ↓ | ↓ | 0 | < 3,5 | < 1 | |
| δβ-talasemia | ↓ | ↓ | 0 | < 3,5 | 5-20 |
Hb: hemoglobina; VCM: volumen corpuscular medio.
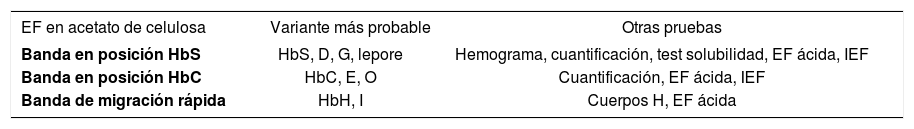
Sugerencias en el diagnóstico diferencial de variantes estructurales
| EF en acetato de celulosa | Variante más probable | Otras pruebas |
|---|---|---|
| Banda en posición HbS | HbS, D, G, lepore | Hemograma, cuantificación, test solubilidad, EF ácida, IEF |
| Banda en posición HbC | HbC, E, O | Cuantificación, EF ácida, IEF |
| Banda de migración rápida | HbH, I | Cuerpos H, EF ácida |
EF: electroforesis; Hb: hemoglobina; IEF: isoelectroenfoque.


