





Puntos clave
- •
Los hidratos de carbono (HC) constituyen la fuente energética cuantitativamente más importante de la dieta.
- •
El proceso de digestión y absorción de los HC es relativamente simple y los trastornos relacionados con este proceso son, en general, de diagnóstico sencillo y tratamiento muy eficaz.
- •
El síntoma más común y característico, sobre todo en el lactante, es la existencia de diarrea con deposiciones numerosas, acuosas, explosivas y de olor ácido, y, en niños mayores, el dolor y la distensión abdominal.
- •
El método diagnóstico más útil es la prueba de absorción oral con determinación de H2 espirado. La determinación simultánea de metano mejora la sensibilidad.
- •
El trastorno más frecuente es el déficit de lactasa de tipo adulto que afecta al 70–75% de la población mundial y está estrechamente relacionado con factores étnicos.
- •
El tratamiento habitual es la reducción más o menos completa de la ingestión del HC implicado.
Los hidratos de carbono (HC) constituyen la fuente energética cuantitativamente más importante de la dieta. Están presentes tanto en tejidos vegetales como animales, en forma de mono, di, oligo y polisacáridos. Un adulto ingiere diariamente 300–400g de HC, que representan el 50–60% de las calorías aportadas por la alimentación. Un niño de 3–5 años ingiere 150–200g/día. En una alimentación variada, estos HC están constituidos en un 50–60% por almidón, 30–40% por sacarosa y el resto por lactosa. La lactosa es el único azúcar de la alimentación del lactante pequeño y cubre aproximadamente el 40% de las necesidades energéticas del niño alimentado al pecho. El proceso de digestión y absorción de los HC es relativamente simple y los trastornos relacionados con este proceso, ya sean primarios o secundarios, constituyen una causa frecuente de diarrea crónica y otros síntomas. Se trata, en general, de trastornos de diagnóstico sencillo y tratamiento muy eficaz.
Lectura rápida
Los hidratos de carbono (HC) constituyen la fuente energética cuantitativamente más importante de la dieta. Los HC más importantes son el almidón, sacarosa y lactosa.
El proceso de digestión y absorción de los HC es relativamente simple y consta de la digestión luminal del almidón, la digestión parietal de los di y oligosacáridos por las oligosacaridasas parietales y la absorción de los monosacáridos (glucosa, galactosa y fructosa) a través de la membrana del borde en cepillo del enterocito.
La malabsorción de HC hace referencia a un fracaso en la digestión o absorción normal de HC y puede acompañarse o no de síntomas y signos de intolerancia clínica. Por el contrario, la intolerancia es la existencia de síntomas como flatulencia, borborigmos, distensión abdominal, abdominalgia o diarrea en relación con la malabsorción de HC.
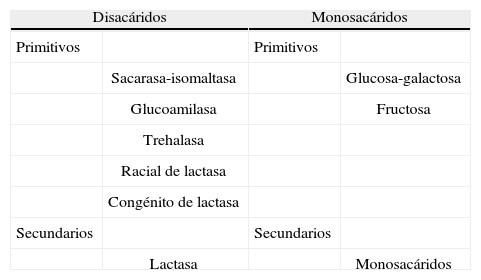
Los trastornos de digestión y absorción de HC pueden clasificarse en primitivos y secundarios y, dependiendo del tipo de HC, en di u oligosacáridos o monosacáridos. Dentro de los trastornos primitivos se incluyen: déficit de sacarasa-isomaltasa, déficit de glucoamilasa, déficit de trehalasa, déficit congénito de lactasa, déficit racial o de tipo adulto de lactasa, malabsorción congénita de glucosa-galactosa y malabsorción de fructosa.
El síntoma más común y característico es la existencia de diarrea con deposiciones numerosas, acuosas y explosivas, que se acompañan de la emisión de ruidos hidroaéreos y producen eritema perianal. Otros síntomas y signos acompañantes son distensión abdominal, borborigmos y flatulencia. También son frecuentes la irritabilidad, el dolor abdominal, la deshidratación y el fracaso de desarrollo.
El diagnóstico de malabsorción de HC se basa en la sospecha clínica y la determinación del pH fecal y la presencia de cuerpos reductores en heces. Para determinar la absorción y la tolerancia a un HC, se utiliza la prueba de absorción oral. Es aconsejable complementarla con una prueba de H2 espirado. En caso de sospecha de déficit primitivo, pueden determinarse las oligosacaridasas parietales (lactasa, sacarasa y maltasa) y realizar un estudio genético. Por último, la confirmación diagnóstica exige la normalización clínica tras la retirada de la dieta del HC sospechoso.
El déficit congénito de sacarasa-isomaltasa es un trastorno de herencia autosómica recesiva. El cuadro se inicia a la 6–18 meses tras la introducción de sacarosa y almidón en la dieta. El diagnóstico se basa en la sospecha clínica y el test de H2 alterado tras administración de sacarosa y en la determinación de la actividad oligosacaridásica parietal. El tratamiento se basa en la restricción de la ingesta de sacarosa y almidón. Los déficits de glucoamilasa y trehalasa son raros.
El déficit de lactasa es el trastorno más frecuente. Aproximadamente, el 70–75% de la población mundial tiene deficiencia de lactasa. La prevalencia de persistencia/no persistencia de lactasa está estrechamente relacionada con la raza. En el déficit racial, el descenso se inicia de manera paulatina después de los 5 años hasta alcanzar una actividad residual del 10–25% en la adolescencia.
El déficit secundario de lactasa implica la existencia de un trastorno subyacente responsable de la deficiencia de lactasa y de la consiguiente malabsorción de lactosa. La causa más frecuente es la injuria infecciosa. Es de peor pronóstico en menores de 3 meses y malnutridos y el trastorno es siempre transitorio.
El déficit congénito de lactasa es un trastorno extremadamente raro. Los neonatos afectados presentan diarrea grave con deshidratación al introducir lactosa en la dieta. El diagnóstico se confirma al demostrar ausencia de lactasa en una mucosa yeyunal histológicamente normal. El tratamiento consiste en alimentar con una fórmula libre de lactosa.
Para el diagnóstico de déficit de lactasa raramente son necesarios el estudio genético y la determinación de actividad lactasa. La ingesta de lactosa debe ajustarse a la tolerancia. Habitualmente, el yogur y el queso son bien tolerados.
La malabsorción congénita de glucosa-galactosa es un raro trastorno en el que los pacientes presentan diarrea grave con deshidratación desde los primeros días de vida. La mucosa yeyunal y las oligosacaridasas son normales. El diagnóstico se establece por pruebas de absorción oral con determinación de H2 espirado. La diarrea cesa al eliminar la glucosa y la galactosa de la dieta. El defecto en el transporte se mantiene a lo largo de la vida.
El almidón está formado por polímeros de glucosa de alto peso molecular. Se distinguen 2 tipos: la amilosa, cadena lineal de moléculas de glucosa con enlaces α1–4, y la amilopectina, que, además, presenta enlaces ramificados α1–6 cada 20–25 unidades de glucosa. La proporción entre ambas varía según el origen: 20/80 en arroz y maíz, 60/40 en el trigo y 70/30 en la patata. La digestión de estas macromoléculas se inicia en la boca por la α-amilasa salival, pero la mayor parte es debida a la α-amilasa pancreática en la luz duodenal. Se trata de endoamilasas que rompen los enlaces α1–4 lejos de los extremos de las cadenas y de los puntos de ramificación. Liberan, sobre todo, maltosa (2 moléculas de glucosa), maltotriosa (3 moléculas de glucosa) y residuos de más alto peso molecular (5–10 unidades de glucosa), ramificados si es sustrato es la amilopectina: las dextrinas límite1.
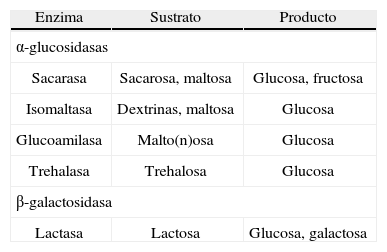
Digestión parietal de di y oligosacáridosAfecta a los productos de digestión del almidón y a los disacáridos naturales: sacarosa (glucosa y fructosa con enlace α1–2), lactosa (glucosa y galactosa con enlace α1–4) y trehalosa (2 moléculas de glucosa con enlace α1–1). Existen 2 grupos de oligosacaridasas: las α-glucosidasas (sacarasa-isomaltasa [SI], glucoamilasa y trehalasa) y la a-galactosidasa neutra (lactasa).
Las 3 oligosacaridasas principales (SI, lactasa y glucoamilasa) son activas en la membrana del borde en cepillo del enterocito. La actividad enzimática es nula en el fondo de la cripta, aparece en el tercio inferior de la vellosidad, es máxima en el tercio medio y comienza a declinar en el tercio superior, donde los enterocitos descaman. Las actividades SI y lactasa son nulas en el píloro, aumentan a lo largo del duodeno para alcanzar su máxima actividad en las primeras asas yeyunales y decrecer en el íleon. La actividad glucoamilasa, por el contrario, aumenta a lo largo del intestino y es máxima en el íleon terminal.
La SI es responsable del 75–80% de la actividad maltásica total de la mucosa intestinal, de la casi totalidad de la actividad isomaltásica y de la totalidad de la actividad sacarásica. La sacarasa hidroliza la sacarosa, maltosa y maltotriosa, y la isomaltasa, las dextrinas, maltosa y maltotriosa. Ninguna de ellas puede hidrolizar oligosacáridos de más de 4 unidades de glucosa. La actividad SI es inducible por la ingestión de sacarosa y fructosa (tabla 1).
La glucoamilasa es responsable del 20–25% de la actividad maltásica total y de la totalidad de la activad sobre los oligómeros de glucosa de más de 4 unidades. Su actividad es mayor cuanto más larga sea la cadena de glucosa.
La lactasa es responsable de la práctica totalidad de la actividad intestinal sobre la lactosa. El resultado final de la acción de las oligosacaridasas son los monosacáridos (glucosa, galactosa y fructosa), única forma en que la mucosa intestinal absorbe los HC.
Absorción de monosacáridosLos monosacáridos no pueden difundir a través de las fases lipídicas de las membranas de los enterocitos ni atravesar las uniones intercelulares, de forma que deben utilizar sistemas de transporte específicos. Los transportadores están situados en la membrana del borde en cepillo y, por su naturaleza, son saturables.
Transporte de glucosa-galactosaEs el sistema de transporte más importante, ya que asegura la absorción de más del 80% de los HC, es decir, la mitad de la energía aportada por la alimentación y es de 2 a 3 veces más eficaz en yeyuno que en íleon. El transportador más importante y conocido es el SGLT1, que es el responsable del transporte activo de glucosa a través de la membrana en cepillo del enterocito. El SGLT1 asocia el transporte de 2 iones de sodio y una molécula de glucosa. El gradiente de sodio intracelular se mantiene gracias a la ATPasa Na+/K+. Esta bomba Na+/K+ permite el transporte activo de sodio desde en interior del enterocito hacia la sangre a través de la membrana basolateral. Se trata, pues, de un transporte Na+ dependiente, indirectamente activo. La glucosa acumulada en el enterocito pasa a la sangre por exocitosis o por transporte pasivo a través de la membrana basolateral mediante el transportador GLUT2. Es un sistema saturable y sometido a inhibición competitiva2,3.
Transporte de fructosaLa fructosa es absorbida pasivamente mediante un proceso absolutamente independiente de la absorción de glucosa. Utiliza transportadores del grupo GLUT (fundamentalmente GLUT2 y GLUT5), aún no completamente conocidos. Parece que las vías GLUT8 y GLUT12 constituyen mecanismos regulatorios adaptativos para acomodarse a grandes ingestas de fructosa. La capacidad de absorción de fructosa es limitada, de forma que la ingestión de 50g de fructosa produce síntomas de intolerancia en el 70% de los adultos jóvenes sanos.
Rescate colónicoEn población sana, entre un 2–20% del almidón ingerido alcanza el colon sin ser absorbido en intestino delgado. Los HC no pueden ser absorbidos por la mucosa colónica, pero sí pueden ser metabolizados por la flora bacteriana. El metabolismo bacteriano anaeróbico produce mono y disacáridos que son metabolizados con producción de ácido láctico y ácidos grasos de cadena corta (acetato, propionato, butirato) y gases (hidrógeno, metano, dióxido de carbono). Una proporción considerable (hasta el 90%) de estos ácidos orgánicos y gases son absorbidos por la mucosa colónica con el consiguiente aprovechamiento energético (entre 3,5–5,9 Kcal/g)4.
Malabsorción e intolerancia a hidratos de carbonoMalabsorción e intolerancia no son términos sinónimos. La malabsorción de HC hace referencia a un fracaso en la digestión o absorción normal de HC, que permite que estos alcancen el colon y puede acompañarse o no de síntomas y signos de intolerancia clínica. Por el contrario, la intolerancia es la existencia de síntomas como flatulencia, borborigmos, distensión abdominal, abdominalgia o diarrea en relación a la malabsorción de HC4. Los síntomas de intolerancia a HC pueden ser el resultado de:
- 1.
Defectos congénitos o adquiridos de la secreción pancreática exocrina o de las oligosacaridasas parietales.
- 2.
Disminución de la absorción de monosacáridos por defectos de los mecanismos de transporte o reducción de la superficie absortiva.
- 3.
Ingestión excesiva de HC para los que fisiológicamente el intestino humano tiene una capacidad absortiva limitada, como fructosa, sorbitol y manitol.
- 4.
Administración terapéutica de HC no absorbibles (lactulosa, lactitiol), inhibidores de la absorción de HC (acarbosa, metformina) o sucedáneos dulces (sorbitol, fructosa).
- 5.
Antibióticos que interfieren con el rescate colónico de los HC no absorbidos en el intestino delgado.
Los trastornos de digestión y absorción de HC pueden clasificarse en primitivos y secundarios y, dependiendo del tipo de HC, en di u oligosacáridos o monosacáridos (tabla 2). Dentro de los trastornos primitivos se incluyen: déficit de SI (DCSI), déficit de glucoamilasa, déficit de trehalasa, déficit congénito de lactasa, déficit racial o de tipo adulto de lactasa, malabsorción congénita de glucosa-galactosa y malabsorción de fructosa. El trastorno secundario más frecuente es la intolerancia transitoria a la lactosa.
FisiopatologíaLa maldigestión o malabsorción produce un acúmulo de HC en el intestino que produce un efecto osmótico con atracción de agua y electrolitos hacia la luz intestinal. El aumento del volumen intraluminal provoca un aumento del peristaltismo y un aumento del flujo ileal. Los HC no digeridos o absorbidos alcanzan el colon, donde son fermentados por la flora cólica con producción de ácido láctico, ácidos grasos de cadena corta y gases. El resultado final es la existencia de meteorismo y diarrea acuosa y ácida4.
ClínicaTodos los trastornos de intolerancia a HC tienen una sintomatología clínica común. Las diferencias se establecen en cuanto al momento y la edad de aparición de los síntomas y su relación con la ingestión de un HC concreto. El síntoma más común y característico, sobre todo en el lactante, es la existencia de diarrea con deposiciones numerosas, acuosas y explosivas, de olor ácido, que se acompañan de la emisión de ruidos hidroaéreos y producen eritema perianal. Otros síntomas y signos acompañantes son distensión abdominal, borborigmos y flatulencia. También son muy frecuentes la irritabilidad en el lactante y el dolor abdominal por distensión gaseosa e hiperperistaltismo4. En el lactante, puede producirse deshidratación por pérdida hídrica y fracaso de desarrollo por la pérdida energética de los HC no absorbidos y la malabsorción grasa por maldigestión, relacionada con la incapacidad de las sales biliares para alcanzar una concentración micelar crítica por la dilución secundaria al aumento de volumen intraluminal.
DiagnósticoEl diagnóstico de malabsorción de HC requiere una serie de pasos escalonados hasta la valoración de la respuesta terapéutica (tabla 3). La sospecha clínica se basa en el desarrollo de los síntomas y signos descritos, tras la ingestión de un HC determinado. El primer paso de la confirmación diagnóstica es la determinación del pH fecal y de la presencia de cuerpos reductores en heces mientras el paciente recibe el HC a investigar. El pH fecal normal es mayor de 5,5, excepto en el lactante, que recibe leche materna. El pH fecal ácido indica fermentación de HC por la flora cólica y producción de ácido láctico y otros ácidos orgánicos. La determinación de cuerpos reductores es una prueba semicuantitativa y en la valoración debe tenerse en cuenta que la sacarosa es un azúcar no reductor.
Para determinar la absorción y tolerancia a un HC concreto, se utilizan las pruebas de absorción oral. La prueba consiste en la administración del HC a investigar a dosis de 1–2g/kg (máximo 25–50g) en solución al 10–20%, para evitar el efecto osmótico, con valoración de la respuesta clínica y determinación de glucemia cada 30min. Se considera normal un ascenso de la glucemia superior a 20–25mg/dl. La prueba tiene un elevado porcentaje de resultados falsos positivos y negativos, por lo que es aconsejable complementarla con una prueba de H2 espirado que informa sobre la presencia en la luz de un HC que ha sido fermentado por la flora intestinal con producción de gas. El H2 se determina por cromatografía de gas cada 30min durante 210min y se considera positivo un ascenso mayor de 20ppm. La sensibilidad aumenta si se determina simultáneamente metano y puede dar resultados falsamente negativos en caso de ausencia de flora fermentadora. El resultado también puede alterarse por el empleo de antibióticos y laxantes. En caso de resultado positivo al investigar un disacárido, puede repetirse la prueba utilizando los monosacáridos correspondientes para discriminar si se trata de un trastorno en la hidrólisis del disacárido o en el transporte de los monosacáridos5.
En caso de sospecha de déficit primitivo, pueden determinarse las oligosacaridasas parietales, preferiblemente en una muestra obtenida en el ángulo de Treitz y siempre en una mucosa yeyunal histológicamente normal6. Habitualmente, se determinan las actividades lactasa, sacarasa y maltasa. En los tipos primitivos puede realizarse un estudio genético en la búsqueda de las mutaciones conocidas de la enfermedad. Por último, la confirmación diagnóstica exige la normalización clínica tras la retirada de la dieta del HC sospechoso.
Trastornos específicosDéficit congénito de sacarasa-isomaltasaEs un trastorno de herencia autosómica recesiva, en el que existe un defecto en la hidrólisis de la sacarosa, maltosa, dextrinas y almidón por una deficiencia en el complejo SI. La ingestión de estos nutrientes provoca una diarrea osmótica con dolor y distensión abdominal y, a medio plazo, malnutrición y fracaso de desarrollo. El gen de la SI está localizado en el cromosoma 3 y hasta el momento se han descrito más de 25 mutaciones. La heterogeneidad fenotípica intracelular se refleja en grandes variaciones de la capacidad enzimática residual, desde absolutamente ausente hasta una disminución moderada, y se relaciona con la gravedad de las manifestaciones clínicas. La prevalencia de DCSI es objeto de debate por la posibilidad de infradiagnóstico7. Se estima en un 5–10% en esquimales de Groenlandia, 3–7% en población nativa de Canadá y 3% en nativos de Alaska. En población norteamericana blanca no hispánica, la prevalencia varía entre 1/500 y 1/2.000, y es menor en afroamericanos e hispánicos. Son frecuentes los heterocigotos con actividad enzimática disminuida. El cuadro se inicia a la 6–18 meses tras la introducción de sacarosa y almidón en la dieta. También puede manifestarse en forma de dolor abdominal recurrente, colon irritable o dispepsia8.
El diagnóstico se basa en la sospecha clínica y el test de H2 alterado tras administración de sacarosa y en la determinación de la actividad oligosacaridásica parietal que muestra una disminución o ausencia de la actividad sacarasa (razón sacarasa/lactasa < 1), moderada disminución de la actividad maltasa y actividad lactasa normal. Pueden investigarse mutaciones del gen SI, pudiendo encontrar alguna de las 4 más frecuentes en el 83% de los pacientes9. El tratamiento se basa en la restricción de la ingesta de sacarosa y almidón10. En general, la tolerancia va mejorando con la edad por el aumento de la longitud del intestino delgado y la mejora del rescate colónico de HC. Habitualmente, el almidón es bien tolerado a partir del año de edad. En casos graves, puede administrarse la enzima sacarosidasa, obtenida de Saccharomyces cerevisiae11,12.
Déficit de glucoamilasaEs un trastorno descrito en 1994, caracterizado por intolerancia clínica tras la ingestión de almidón y polímeros de glucosa, debido a una disminución de la actividad de la glucoamilasa13. Puede tratarse de un trastorno primitivo o secundario. Los síntomas son los habituales en todas las intolerancias a HC. En los casos primitivos, el trastorno es permanente. El diagnóstico se basa en la prueba de H2 tras la administración de almidón y en la determinación de la actividad glucoamilasa parietal. En algún caso puede asociarse a otras deficiencias enzimáticas14. El tratamiento consiste en la restricción de almidón y polímeros de glucosa.
Déficit de trehalasaLos pacientes afectados de este trastorno presentan vómitos, dolor abdominal y diarrea tras la ingestión de setas15,16. El diagnóstico se basa en la clínica y la determinación de la actividad trehalasa en la mucosa intestinal. El tratamiento es la dieta de exclusión.
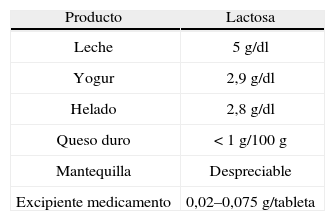
Déficit de lactasaLa lactosa es el HC de la leche de los mamíferos, por ello, la mayor fuente de lactosa son la leche y los derivados lácteos. La concentración de lactosa en la leche varía desde indicios en las focas y leones marinos hasta los 7,5g/dl de los humanos. Las leches de vaca, cabra y oveja contienen aproximadamente 5g/dl (tabla 4). La gravedad de los síntomas depende de la cantidad de lactosa ingerida y de la actividad lactásica intestinal. Se trata, con mucha diferencia, del trastorno de los HC más frecuente17,18.
También denominado déficit primitivo de lactasa, hipolactasia de tipo adulto, lactasa no persistente o déficit hereditario de lactasa. Aproximadamente, el 70–75% de la población mundial tiene deficiencia de lactasa. La prevalencia de persistencia/no persistencia de lactasa está estrechamente relacionada con la raza. La mayoría de los adultos de Asia y poblaciones nativas de Norteamérica (100%), nativos de Latinoamérica (50–80%) y negros africanos y judíos asquenazíes (60–80%) son lactasa no persistentes, mientras que la mayoría de los blancos son lactasa persistentes. Además, existen grandes diferencias entre blancos del norte y sur de Europa. En el norte de Europa, la prevalencia de deficiencia de lactasa es inferior al 2%, ascendiendo hasta un 20–25% en el sur (Grecia, sur de Italia, España). El descenso de la actividad lactasa es más precoz (2 años) en asiáticos con lactasa no persistente que en poblaciones blancas con alta prevalencia de lactasa persistente (10–20 años de edad en finlandeses). En general, el descenso se inicia de manera paulatina después de los 5 años, hasta alcanzar una actividad residual del 10–25% en la adolescencia.
La síntesis de lactasa es una función del gen LCT, localizado en el brazo largo del cromosoma 2. La expresión de LCT es regulada por un promotor localizado más arriba del gen LCT y la correlación entre raza y persistencia/no persistencia de lactasa está ligada a variaciones genéticas del promotor de lactasa, no del gen LCT. El promotor natural observado en la mayoría de los humanos con lactasa no persistente contiene citosina/citosina en la posición 13910 (C/C-13910). En las poblaciones del norte de Europa con alta prevalencia de lactasa persistente, una o 2 citosinas han sido sustituidas por timina (T/C o T/T-13910). Estas mutaciones eliminan la reducción programada de lactasa con la edad, manteniendo niveles infantiles de lactasa en la edad adulta19,20.
Déficit secundario de lactasaImplica la existencia de un trastorno subyacente responsable de la deficiencia de lactasa y de la consiguiente malabsorción de lactosa. La causa más frecuente es la injuria infecciosa (p. ej., rotavirus) que produce pérdida de los enterocitos de la parte alta de la vellosidad que son sustituidos por enterocitos inmaduros con bajo contenido en lactasa. Otras posibles causas de deficiencia secundaria de lactasa son las infestaciones parasitarias (Giardia, Criptosporidium), enfermedad celíaca, enfermedad de Crohn, inmunodeficiencias, alergia a proteínas de leche de vaca y cirugía intestinal. Es más frecuente y de peor pronóstico en menores de 3 meses y malnutridos. El trastorno es siempre transitorio y, con frecuencia, no requiere la retirada de la lactosa de la dieta.
Déficit madurativo de lactasaLa lactasa es deficiente hasta la 34.a semana de gestación. Los lactantes pretérmino pueden beneficiarse del uso de fórmulas suplementadas con lactasa o reducidas en lactosa21, pero, con frecuencia, la leche de mujer no tiene efectos perjudiciales a corto y largo plazo. En el neonato, más de un 20% de la lactosa ingerida puede alcanzar el colon, produciendo un pH fecal ácido (5–5,5) y favoreciendo el desarrollo de ciertas especies bacterianas (Bifidobacterium y Lactobacillus).
Déficit congénito de lactasaEs un trastorno extremadamente raro22. Antes del siglo XX, los niños con deficiencia congénita de lactasa no podían sobrevivir dada la inexistencia de sustitutos lácteos accesibles y nutricionalmente adecuados. Los neonatos afectados presentan diarrea grave, con deshidratación al introducir lactosa en la dieta. El diagnóstico se confirma al demostrar ausencia de lactasa en una mucosa yeyunal histológicamente normal. El tratamiento consiste en alimentar con una fórmula libre de lactosa.
El diagnóstico de intolerancia a la lactosa debe sospecharse por la clínica, apoyarse en el pH fecal ácido y los cuerpos reductores en heces y en la respuesta a la dieta de exclusión y confirmarse, aunque no siempre es necesario, con la prueba de H2 espirado. Raramente, son necesarios el estudio genético y la determinación de actividad lactasa. La ingesta de lactosa debe ajustarse a la tolerancia. Pueden utilizarse fórmulas bajas o libres de lactosa y, habitualmente, el yogur y el queso son bien tolerados. Los medicamentos que contienen lactosa como excipiente raramente tienen relevancia clínica23.
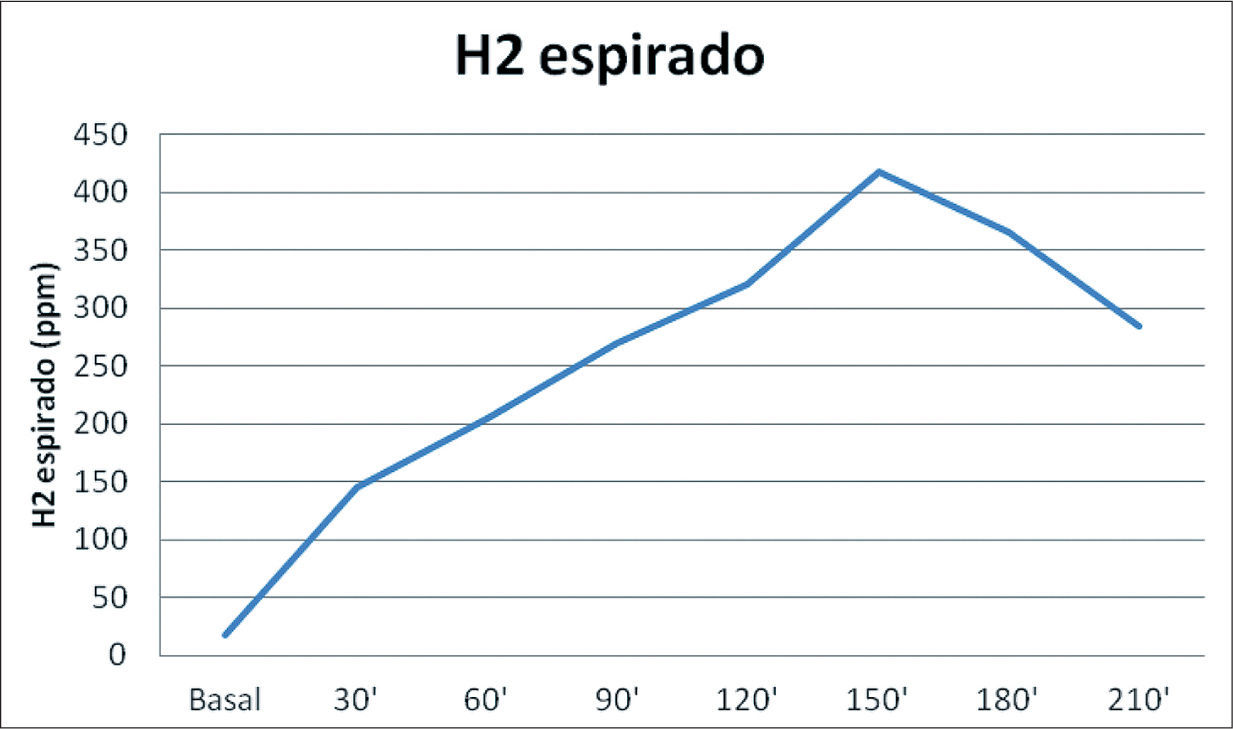
Malabsorción congénita de glucosa-galactosaEs un raro trastorno congénito de herencia autosómica recesiva, en el que existe un defecto en el transportador SLGT1 por mutaciones en el gen SLC5A124. Los pacientes con este defecto presentan diarrea grave con deshidratación desde los primeros días de vida. La mucosa yeyunal y las oligosacaridasas son normales. El diagnóstico se establece por pruebas de absorción oral con determinación de H2 espirado (fig. 1). La diarrea cesa al eliminar la glucosa y la galactosa de la dieta. Después de los 3 meses pueden tolerarse pequeñas cantidades de glucosa y galactosa25, pero el defecto en el transporte se mantiene a lo largo de la vida.
Malabsorción de fructosa
La fructosa se encuentra en la dieta en forma de disacárido como sacarosa o de monosacárido como edulcorante de muchos alimentos. La capacidad de absorción de fructosa es limitada, sobre todo, si no va acompañada por glucosa, de forma que la ingestión de 50g de fructosa produce síntomas de intolerancia en adultos sanos. La eficiencia de la absorción de fructosa puede modularse por la coingestión de otros azúcares y polioles26. La ingestión de alimentos ricos en fructosa puede producir distensión abdominal y diarrea, y provocar síntomas en pacientes con síndrome de colon irritable y puede ser causa de dolor abdominal funcional en niños27. La prueba de H2 espirado puede ser útil para identificar a los pacientes que pueden beneficiarse de una dieta restringida en fructosa. La restricción de fructosa puede ser un tratamiento efectivo de los síntomas intestinales funcionales, pero puede tener un impacto negativo sobre la flora colónica.