















El síndrome hemolítico urémico (SHU) es una de las causas más frecuentes de insuficiencia renal aguda (IRA) en pediatría y es la causa del 4,5% de los casos de niños con insuficiencia renal crónica (IRC) en tratamiento con depuración extrarrenal. Se caracteriza por la presencia simultánea de anemia hemolítica microangiopática, trombocitopenia e IRA, con afectación variable de otros órganos (sistema nervioso central [SNC], respiratorio, hígado, páncreas, etc.)1–4.
Existe controversia en su relación con la púrpura trombótica trombocitopénica (PTT), y comparte con ella manifestaciones clínicas, fisiopatológicas y anatomopatológicas, aunque la etiología puede ser diferente. Se denomina SHU a la microangiopatía que afecta preferentemente al riñón, siendo el cuadro clínico más frecuentemente observado en la infancia y PTT cuando la microangiopatía trombótica afecta fundamentalmente al SNC, proceso muy poco frecuente en niños5–7.
Puntos clave

El SHU se ha clasificado históricamente en 2 tipos: a) asociado a diarrea o SHU típico (90% de los casos)1,6, o b) no asociado a diarrea (SHU atípico). El SHU típico se produce en la mayoría de los casos por infección por bacterias gastrointestinales productoras de verocitotoxinas o toxinas shiga-like (Stx), de las que Escherichia coli enterohemorrágica (ECEH o STEC)8 es la más frecuentemente implicada en el ámbito mundial y Shigella disenteriae9 en algunas regiones tropicales. Se han descrito también casos de SHU por Citrobacter freundii.
El SHU atípico representa el 10% de los casos y puede tener distintas etiologías: infección por Streptococcus pneumoniae (40% de los casos no asociados a ECEH), alteraciones genéticas o adquiridas en la regulación de la activación del complemento, fármacos, etc.
El avance en el conocimiento de la etiología y su importancia en el tratamiento y pronóstico de estos pacientes ha llevado al establecimiento de una nueva clasificación en 2 grupos (tabla 1), según se conozca la etiología precisa o no5.
Clasificación etiológica del síndrome hemolítico urémico
| Categoría | Características |
|---|---|
| Nivel 1 | Etiología conocida |
| 1.1. | Inducido por infección
|
| 1.2. | Alteraciones en la regulación del complemento
|
| 1.3. | Deficiencia de la proteasa del factor de Von Willebrand, ADAMTS 13
|
| 1.4. | Alteración en el metabolismo de la cobalamina |
| 1.5. | Inducida por quinina |
| Nivel 2 | Etiología desconocida. Asociado a: |
| 2.1. | Infección por virus de la inmunodeficiencia humana |
| 2.1. | Enfermedades malignas, quimioterapia y radiaciones ionizantes |
| 2.1. | Inhibidores de la calcineurina (ciclosporina, tacrolimus) y trasplante |
| 2.1. | Embarazo, síndrome HELLP, anticonceptivos |
| 2.1. | Lupus eritematoso diseminado y síndrome antifosfolipídico |
| 2.1. | Glomerulopatías |
| 2.1. | Familiar, no incluido en nivel 1 |
| 2.1. | Inclasificado |
HELLP: hemólisis, enzimas hepáticas elevadas y trombocitopenia.
En general, la edad y la forma de presentación clínica orientan el diagnóstico etiológico. Sin embargo, es importante recalcar que un mismo paciente puede presentar factores etiológicos asociados, de tal modo que un paciente con un factor predisponente, como es la alteración de la regulación del complemento, puede desarrollar el SHU tras un factor desencadenante como la infección5.
Síndrome hemolítico urémico asociado a verocitotoxina o toxina shiga-likeEs el SHU clásico, típico o posdiarreico (SHU D+)5,6 secundario a infección por ECEH. Se caracteriza por aparición de anemia, trombocitopenia y daño renal agudo tras un cuadro de diarrea con sangre y dolor abdominal. Además, otras infecciones provocadas por ECEH como las del tracto urinario pueden seguirse de SHU, sin pródromo de diarrea, aunque con fisiopatología, clínica y pronóstico similares.


El SHU producido por Stx es más frecuente en niños menores de 5 años. El vector principal es el ganado vacuno y la infección ocurre por consumo de carne poco cocinada, lácteos no pasteurizados, frutas o vegetales. La mayor incidencia es en los meses de verano de forma esporádica, aunque se han descrito epidemias en el contexto de aguas o comida contaminadas. La transmisión persona a persona también es posible. E. coli O157:H7 es el serotipo que con más frecuencia provoca SHU D+; sin embargo, en los últimos años han emergido otros serotipos no –O157:H7 (O26:H11/H –, O157: H -, etc.). La probabilidad de desarrollar SHU después de la infección depende del serotipo implicado, siendo para O157:H7 del 15%10–12. Estudios recientes refieren más probabilidad en los pacientes infectados por O157: H – y menor para no -O157:H712. El uso de fármacos inhibidores de la motilidad intestinal se ha asociado a un riesgo mayor de SHU12–14. En un metaanálisis reciente no se ha confirmado el incremento del riesgo en relación con el uso de antibióticos15. La administración de fluidoterapia agresiva en la fase de diarrea se asocia a evolución más leve del SHU y una tendencia menor a oligoanuria16. Se ha indicado que alteraciones en la regulación del complemento pueden favorecer el desarrollo de SHU D+.
PatogeniaEl conocimiento de la fisiopatología del SHU permite comprender los mecanismos precisos de daño celular y elegir tratamientos específicos, así como estrategias preventivas.
Distintos factores de virulencia de ECEH están implicados en la capacidad de colonizar el intestino humano, adherirse a la mucosa, dañar microvellosidades y liberar citotoxinas, con lo que se contribuye a las manifestaciones de la diarrea con sangre, y a la lesión endotelial con trombosis secundaria que ocurre en arteriolas y glomérulos renales. La capacidad para excretar Stx es el requerimiento principal de ECEH o de Shigella para causar SHU, ya que las toxinas son principales factores en la patogenia del daño del endotelio microvascular que ocurre en el SHU. Están codificadas en un bacteriófago insertado en el cromosoma bacteriano. Su estructura consta de una subunidad A central y 5 subunidades B periféricas. Hay 2 tipos de toxina shiga: Stx 1 y Stx 2. La Stx 1 difiere tan sólo en un aminoácido de la toxina shiga de Shigella dysenteriae tipo 1. La Stx 2 presenta varios subtipos. Se ha demostrado que las cepas de ECEH productoras de determinados subtipos de Stx 2 presentan cuadros más graves que las productoras de Stx 117,18.
La capacidad de adherencia y fijación íntima a la superficie celular del enterocito que provoca pérdida de microvellosidades es uno de los factores de más virulencia, ya que permite la secreción de las Stx muy próximas a los enterocitos y facilita su paso a la sangre por translocación a través del epitelio intestinal. Las toxinas circulantes se fijan a las células diana mediante la interacción de las subunidades B con un receptor específico de la membrana de estas células, el glucoesfingolípido globotriosil cerámida (Gb3). La toxina es entonces endocitada y la subunidad A produce el bloqueo de la síntesis proteica y la muerte celular. La susceptibilidad de un tipo celular depende de la cantidad de receptores para toxina que exprese y de la estructura de su porción lipídica. Esto explica por qué algunas líneas celulares son refractarias a las toxinas, limitando el daño a los órganos susceptibles.
La interacción entre las Stx y las citocinas incrementa el daño. Las Stx estimulan la activación y la migración de los leucocitos4, así como la producción de citocinas y otros mediadores de la inflamación, lo que potencia su toxicidad. A su vez, las citocinas aumentan la expresión de receptores Gb3 para toxina en las células endoteliales amplificando su efecto y provocando lesiones histopatológicas más graves13. El daño del endotelio favorece la agregación plaquetaria y el depósito de fibrina, el cual provoca microtrombos. Estos ocluyen la luz de los vasos y generan anemia hemolítica microangiopática y trombocitopenia. Además, hay un daño directo en las membranas de los hematíes y un estado de activación de las plaquetas que acelera su destrucción. El daño renal no sólo se limita a la afectación glomerular y arteriolar, sino que se ha comprobado también lesión de células tubulares renales y mesangiales19.
Lectura rápida
La etiología en los niños es diversa: en el 90% de los casos se debe a infección por Escherichia coli enterohemorrágica o Shigella disenteriae (SHU típico), en el 5% de los casos es secundaria a infección invasiva por Streptococcus pneumoniae y en menos del 5%, por alteraciones en las proteínas reguladoras del complemento, ADAMTS 13 o en el metabolismo de la cobalamina. Puede presentarse en pacientes trasplantados especialmente de médula ósea, en asociación con infección por el virus de la inmunodeficiencia humana o en pacientes tratados con quimioterápicos o inmunodepresores.
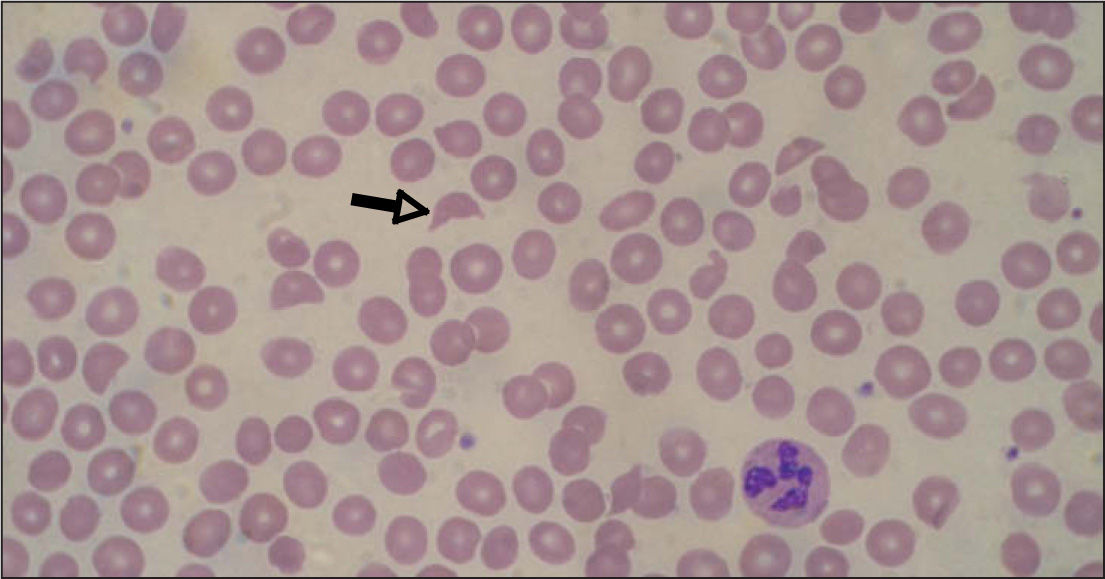
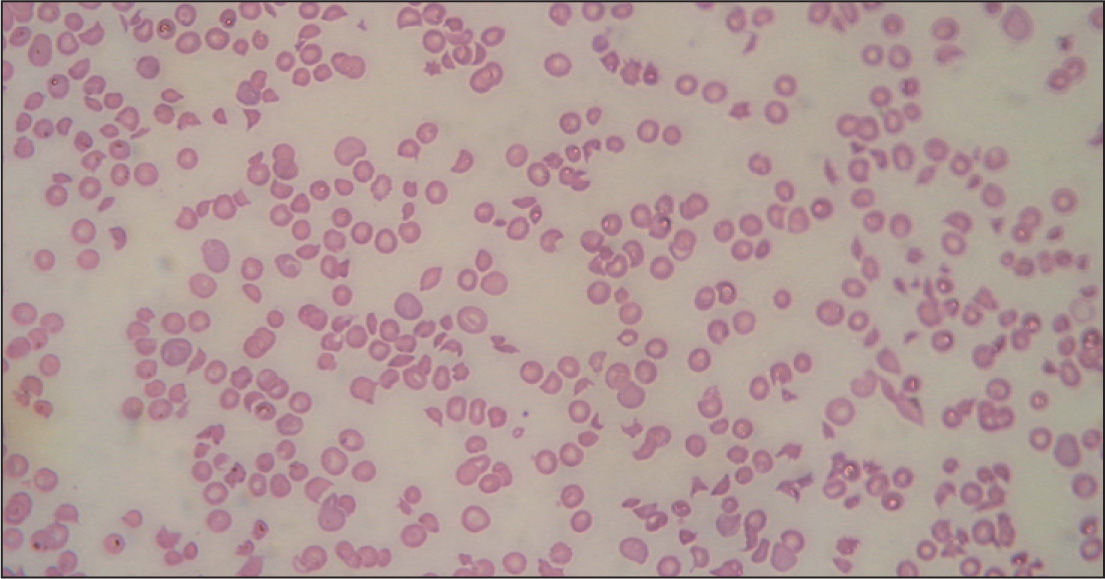
Inicialmente, los pacientes desarrollan un cuadro de diarrea acuosa seguida de diarrea sanguinolenta con dolor abdominal, náuseas y vómitos. La fiebre no siempre está presente (30%). Al cabo de 5–10 días aparecen las alteraciones hematológicas y nefrológicas. La anemia hemolítica microangiopática se caracteriza por cifras de hemoglobina de 6–8g/dl6,20, test de Coombs negativo, hematíes fragmentados (esquistocitos) en sangre periférica (figs. 1 y 2) y datos de hemólisis (aumento de bilirrubina indirecta, disminución de haptoglobina y valores muy elevados de lactatodeshidrogenasa). El grado de anemia no se correlaciona con la gravedad de la enfermedad renal. La trombocitopenia se define como el recuento plaquetario por debajo de 140.000/μl, aunque son frecuentes cifras inferiores a 40.000/μl, a pesar de lo cual no suele haber púrpura ni hemorragia activa. Puede mantenerse durante 2–3 semanas6,21. Tampoco hay correlación entre el grado de trombocitopenia y la lesión renal. El grado de afectación renal es variable, desde tan sólo hematuria y proteinuria, a insuficiencia renal grave con oligoanuria (en un 50%)6. La analítica muestra cifras elevadas de urea, creatinina, potasio, fósforo y ácido úrico (resultado del descenso de filtrado y de la hemólisis). Son frecuentes la hiponatremia y la hipoalbuminemia por hemodilución, la hipocalcemia y la acidosis metabólica. La oligoanuria y la azoemia ocurren en un 50-60% con una duración media de una semana. Hasta un 60% de los pacientes requiere diálisis durante un período de 7–10 días6,20,21. La hipertensión arterial es frecuente, la cual se agrava por la administración excesiva de fluidos o hemoderivados.


Se producen complicaciones neurológicas hasta en un 25%6,21, incluidos alteración en comportamiento, crisis, coma o edema cerebral, que incrementan la mortalidad. En el ámbito digestivo puede producirse colitis hemorrágica grave, necrosis intestinal, prolapso rectal, peritonitis o invaginación. La afectación pancreática leve con intolerancia glucídica es frecuente, aunque ocasionalmente puede complicarse con necrosis o seudoquiste y necesidad de insulinoterapia permanente. Son frecuentes la hepatomegalia o un aumento leve de las transaminasas. Puede aparecer edema pulmonar secundario a sobrecarga hídrica. Las complicaciones cardíacas por isquemia, uremia o sobrecarga volémica son raras, siendo el nivel de troponina I un buen marcador6,12,21,22.
La gravedad del período prodrómico se correlaciona con la gravedad del daño extraintestinal.
Lectura rápida
El SHU típico se presenta habitualmente en los meses de verano; afecta a niños menores de 5 años y es característica la presencia de diarrea sanguinolenta en los días previos al desarrollo de la anemia, trombocitopenia e insuficiencia renal. El SHU asociado a S. pneumoniae se presenta habitualmente asociado a neumonía o meningitis en niños menores de 2 años. Su curso es más grave con más riesgo de complicaciones. El SHU por alteración en las proteínas reguladoras del complemento puede presentarse en los primeros meses de vida, y son frecuentes los antecedentes familiares. Tiene un riesgo de recurrencia elevado.
El diagnóstico de SHU se realiza fundamentalmente mediante datos clínicos y analíticos. Aunque la confirmación de infección por ECEH puede realizarse con coprocultivos, la bacteria sólo está presente en las heces en los primeros días de diarrea y, de estarlo, no siempre se consigue crecimiento. El uso del cultivo agar-MacConkey para observar cepas no fermentadoras de sorbitol (O157:H79) no permite detectar otras cepas con capacidad de producir Stx que quedarían sin diagnosticar23. La determinación de anticuerpos frente a lipopolisacárido de ECEH presenta una sensibilidad y especificidad altas6,24. Las nuevas estrategias de diagnóstico se dirigen a la determinación de Stx en heces, reacción en cadena de la polimerasa e hibridación ADN para detectar el gen Stx. Recientemente, se han comercializado tests rápidos que por inmunoanálisis detectan toxinas en heces y cultivos. Su rapidez y sensibilidad indican que serán el método de cribado de elección.
TratamientoEl tratamiento del SHU es básicamente de soporte.
El tratamiento hídrico requiere una valoración cuidadosa del estado volémico del paciente con balances, peso y seguimiento de constantes. En caso de oligoanuria, se realizará restricción hídrica (aportando pérdidas insensibles y diuresis) hasta la recuperación de la función renal. El uso de diuréticos se reserva para situaciones con afectación cardiovascular y deben suspenderse si no hay respuesta. El tratamiento de las alteraciones electrolíticas y la indicación de técnicas de reemplazo renal son las mismas que en cualquier situación de insuficiencia renal. La elección de la técnica (diálisis peritoneal, hemodiálisis, hemodiafiltración veno-venosa continua) depende de la experiencia del personal y la disponibilidad del centro; en caso de afectación abdominal grave con colitis grave o peritonitis, está contraindicada la diálisis peritoneal.
Para evitar las complicaciones cardiovasculares y pulmonares de la hipervolemia, se transfundirá concentrado de hematíes con cifras de hemoglobina inferiores a 6g/dl, administrándose de forma lenta, sin superar cifras de 8–9g/dl. Se interrumpirá si aparecen signos de sobrecarga vascular.
La transfusión de plaquetas se indica sólo en caso de hemorragia activa o ante procedimientos invasivos, para evitar agravar la microangiopatía trombótica.
La hipertensión arterial se trata con la corrección de la hipervolemia y con fármacos antihipertensivos. Son de elección los antagonistas del calcio y los inhibidores de la enzima de conversión de angiotensina (IECA) se reservan para las secuelas a largo plazo, ya que pueden exacerbar el daño renal agudo por disminución de la perfusión.
En caso de manifestaciones neurológicas graves, serán precisas pruebas de imagen para descartar infarto o hemorragia. Las crisis se tratan con antiepilépticos habituales, teniendo en cuenta que pueden ser secundarias a hipertensión arterial o a alteraciones hidroelectrolíticas6,12.
Deben evitarse los antibióticos durante la fase de diarrea, ya que, aunque no se ha confirmado que favorezcan el desarrollo de SHU, tampoco lo previenen6,12.
El uso de estrategias específicas basadas en la fisiopatología está en continua revisión. En la actualidad no hay evidencia a favor del uso de plasma fresco congelado o de plasmaféresis en este grupo de pacientes con SHU asociado a verocitoxinas. Se recomienda plasmaféresis en pacientes con alteraciones neurológicas graves (basado en los resultados de adultos con PTT12,25,26 o en casos en que se compruebe un déficit en el factor H. No se recomienda el uso de agentes antitrombóticos (urocinasa, heparina), ya que no han demostrado su eficacia en estudios prospectivos controlados19.
Los agentes bloqueadores de Stx en el ámbito intestinal se han probado sin éxito12,25,27. Se está estudiando el uso de anticuerpos monoclonales frente a Stx o análogos de receptor Gb3 para unir e inactivar la toxina.
PronósticoLa anuria prolongada con necesidad de diálisis, las alteraciones neurológicas al diagnóstico y la leucocitosis empeoran el pronóstico6,12,21. La mortalidad es del 3–5%, generalmente asociada a complicaciones extrarrenales. El 5% de los pacientes tendrán secuelas graves (isquemia cerebral, insuficiencia renal terminal [IRT])5,6,12,21,25.
El seguimiento a largo plazo es importante, ya que un 30-50% presentan secuelas como hipertensión arterial, proteinuria o insuficiencia renal. La duración de la oligoanuria y la leucocitosis se relacionan con secuelas tardías En aquellos que evolucionan a un estadio terminal, está indicado el trasplante renal, ya que la recurrencia es muy rara12,25.
Síndrome hemolítico urémico asociado a infección por Streptococcus pneumoniaeEl SHU asociado a infección por S. pneumoniae representa el 0% del total de los casos de SHU no asociados a ECEH28,29. Su incidencia ha aumentado en los últimos años con el incremento de la enfermedad invasiva neumocócica, neumonía, sepsis o meningitis29. Sin embargo, el riesgo de presentar SHU en estos pacientes es bajo: alrededor del 0,4–0,6%, y afecta fundamentalmente a niños menores de 2 años30,31. Los serotipos causantes encontrados son 14, 23F, 6B 19A, 2, 3, 8, 4, 1 y 9 28–30, sin estar por tanto protegidos los vacunados con la vacuna heptavalente.
PatogeniaLa patogenia del SHU asociado a infección por S. pneumoniae se relaciona con la neuraminidasa, una enzima liberada por el neumococo en el plasma. Esta enzima se fija al ácido siálico de las glucoproteínas de la membrana celular (endotelio, hematíes y plaquetas) exponiendo el antígeno Thomsen-Friedenreich (antígeno T) que está normalmente cubierto por este ácido. La interacción del antígeno T expuesto en la membrana de los hematíes, plaquetas y células endoteliales de la microvasculatura renal con los anticuerpos anti-T presentes de forma natural (inmunoglobulina [Ig] M) en el plasma, explicaría la patogenia de la enfermedad32,33 y la positividad del test de Coombs directo, a diferencia de otras formas de SHU en que es negativo34. Sin embargo, debe haber otros mecanismos implicados, porque aunque en la mayoría de los niños con enfermedad invasiva por S. pneumoniae se puede detectar antígeno T, sólo una minoría desarrolla SHU y no en todos los niños con SHU e infección por S. pneumoniae se detecta antígeno T33.
ClínicaLas manifestaciones clínicas iniciales se deben a la propia enfermedad neumocócica. El SHU puede aparecer entre los días 3 y 10 de la evolución de la infección, frecuentemente entre el 7.° y el 9.° día35. Sin embargo, en ocasiones se observa trombopenia, anemia e insuficiencia renal desde el inicio. El curso clínico suele ser más grave que el de los pacientes con SHU típico. En general, presentan más duración de la anuria, más necesidades transfusionales y depuración extrarrenal en un porcentaje más elevado de casos. Son frecuentes las alteraciones extrarrenales: neurológicas, pancreatitis, afectación hepática o intestinal31.
Lectura rápida
Se realiza tras la valoración de la clínica y las alteraciones analíticas que definen el síndrome. El diagnóstico etiológico se realizará dependiendo de la forma de presentación clínica. En los pacientes con episodio de diarrea sanguinolenta, edad mayor de 6 meses y presentación estacional, se investigará la causa de la infección gastrointestinal. Los niños con SHU y sospecha de infección neumocócica invasiva requerirán confirmación bacteriológica o, en su defecto, detección del antígeno T expuesto en la membrana de los hematíes. En los niños con presentación atípica, se investigará la presencia de alteraciones en las proteínas reguladoras del complemento.

La aparición de trombocitopenia, anemia hemolítica y, sobre todo, insuficiencia renal en el curso de la enfermedad neumocócica debe hacer sospechar SHU, sobre todo si hay discrepancia entre la gravedad de las alteraciones analíticas y el estado infeccioso y hemodinámico del paciente. En los pacientes con sepsis/shock séptico asociada a la infección neumocócica invasiva, estas alteraciones analíticas pueden atribuirse de forma errónea a la propia sepsis. En la mayoría de los casos de SHU, los parámetros de coagulación son normales, reflejan la ausencia de coagulación intravascular diseminada (CID) y apoyan el diagnóstico de SHU. Sin embargo, en presencia de sepsis/ shock séptico, puede haber coagulopatía, lo que dificulta el diagnóstico28. La detección de la actividad de neuraminidasa en el plasma mediante la detección del antígeno T expuesto en los hematíes es altamente sensible (90%), pero escasamente específica (49%), ya que se detecta en presencia de infección neumocócica sin desarrollo de SHU33. Lo mismo ocurre con el test de Coombs positivo en el 90% de los pacientes con SHU secundario a S. pneumoniae, pero sin especificidad conocida34.
La biopsia renal con demostración de microangiopatía trombótica no se realiza habitualmente en el período agudo.
En casos de evolución grave, debe valorarse la existencia de factores de susceptibilidad del paciente, como las alteraciones en las proteínas reguladoras del complemento.
Lectura rápida
El tratamiento inicial del SHU es de soporte. Se corregirán las alteraciones hidroelectrolíticas, con especial atención al equilibrio hídrico. Un porcentaje elevado necesitará depuración extrarrenal. Se transfundirá concentrado de hematíes con cifras de hemoglobina inferior a 6g/dl o repercusión hemodinámica. Se realizará transfusión de plaquetas sólo si hay signos de hemorragia activa o ante procedimientos. Frecuentemente precisan tratamiento con fármacos antihipertensivos. En los pacientes con SHU secundario a S. pneumoniae, debe evitarse la transfusión de plasma, ya que puede incrementar la hemólisis. La plasmaféresis está indicada especialmente en los pacientes con alteraciones de las proteínas reguladoras del complemento.
La infección debe tratarse de forma temprana. Dada la gravedad habitual del proceso, está indicada la cefotaxima en la neumonía, y cefotaxima más vancomicina en caso de meningitis hasta conocer la sensibilidad del germen. Deben controlarse los niveles plasmáticos de vancomicina para evitar la toxicidad renal.
El tratamiento es similar al del SHU asociado a E. coli, aunque estos pacientes precisan con más frecuencia depuración extrarrenal y hemoderivados31. Debe evitarse la transfusión de plasma fresco congelado por la posibilidad de presencia de IgM natural que agrave el proceso. Se recomienda el uso de hemoderivados lavados para eliminar el plasma28,29.
PronósticoLos pacientes con SHU asociado a infección por S. pneumoniae presentan peor pronóstico que los pacientes con SHU típico. La mortalidad es más elevada en los niños con meningitis (37-50%), mientras que los pacientes con neumonía presentan una mortalidad similar al del SHU típico36. Hasta el 10% de los pacientes evoluciona a IRT y el 16% mantiene hipertensión arterial e IRC28,31,37.
Síndrome hemolítico urémico secundario a alteraciones en la regulación del complementoEl sistema del complemento es esencial en la respuesta inmunitaria y constituye uno de los mecanismos principales en la defensa contra la infección bacteriana por lisis directa de las membranas celulares bacterianas, opsonización de antígenos o producción de anafilatoxinas que activan la respuesta inflamatoria. Se compone de una serie de glucoproteínas (denominadas por la letra C y un número) que se encuentran en el suero y que se van activando de forma secuencial con el objetivo de destruir la célula diana. Puede activarse por 3 vías: a) la vía clásica; b) la vía de la lectina, o c) la vía alternativa, que convergen en el punto de la activación del factor C3. La vía alternativa está activada de forma continua y produce factor C3b. Éste se fija a las membranas celulares (de una bacteria o de una célula propia) y se une al factor B, el cual se fracciona y se une al factor C3b originando un complejo denominado C3 convertasa C3bBb, que amplifica la ruptura de C3b y origina la formación de un complejo que provoca la lisis de la membrana celular y la destrucción de las bacterias. Un sistema de proteínas solubles y asociadas a la membrana de las células propias controla la activación del factor C3 en el plasma y sobre las membranas celulares del huésped para evitar el daño del propio huésped. Las principales proteínas que intervienen en la regulación del complemento son el factor H, la proteína cofactor de la membrana (MCP), el factor I y el factor B37.
El SHU atípico se produce por alteración de las proteínas reguladoras que conlleva una protección deficiente de la membrana de la célula endotelial frente a la acción lítica del complemento38–40.
Las características clínicas globales del SHU en estos pacientes son: edad temprana de aparición, no necesidad de enfermedad prodrómica, aparición insidiosa, antecedentes familiares, recaídas frecuentes, alta mortalidad y evolución a IRC muy frecuente, existiendo importantes diferencias en los distintos subgrupos38–40. El único factor con valor pronóstico en cuanto a la evolución a IRT o fallecimiento en el primer año de vida es el nivel de creatinina en el primer episodio40.
Factor HEl factor H es una glucoproteína sintetizada en el hígado y es la más importante en la regulación de la activación del complemento por la vía alternativa. Inhibe la formación de C3 convertasa y acelera su destrucción.
El gen que codifica esta proteína se encuentra en el cromososma 1 (1q32). Se han encontrado hasta 100 mutaciones distintas en pacientes con SHU atípico, que originan déficit de factor H o más frecuentemente alteraciones funcionales del factor H con valores normales. No todos los portadores de la mutación desarrollan la enfermedad, la penetrancia es del 50%, probablemente por la presencia de polimorfismos de los genes que codifican estos factores. Además, se han descrito casos de SHU asociado a déficit funcional de factor H adquirido por autoanticuerpos antifactor H38,41.
La edad habitual de presentación es temprana, en los primeros meses de la vida aunque algunos pueden manifestarse más tardíamente. La clínica aparece de forma insidiosa y sin cuadro diarreico previo, aunque en ocasiones se ha podido identificar algún factor desencadenante (infección vías respiratorias, diarrea, etc.). La recurrencia es frecuente39,40.
En estos pacientes está indicada la plasmaféresis si hay alteración funcional y transfusión de plasma en casos de déficit cuantitativo42. El objetivo de la plasmaféresis es eliminar el factor H defectuoso y proporcionar factor H normal. La progresión a IRT es temprana en un porcentaje alto de los casos (hasta el 40% en el primer episodio). Al año del inicio, el 60% de los niños ha fallecido o presenta IRT40. El trasplante renal presenta un 75% de recaídas, con un 80% de pérdida del injerto39,40,43. Se ha preconizado el transplante hepatorrenal como tratamiento potencial de la enfermedad por la síntesis hepática del factor H44. Un programa intensivo de plasmaféresis peritrasplante podría mejorar el resultado. La administración de concentrado de factor H como tratamiento y prevención de la recurrencia en los trasplantes parece ser una opción en un futuro próximo39,40,43.
Proteína cofactor de membranaLas mutaciones en el gen que codifica la MCP, también localizado en el brazo largo del cromosoma 1 (1q32), disminuyen la expresión de esta proteína en las membranas celulares y provocan daño endotelial por activación anómala del complemento. La aparición de la enfermedad es temprana, aunque excepcionalmente antes del año de vida. Puede haber factores infecciosos desencadenantes, e incluso se han descrito casos de SHU asociado a E. coli en pacientes con mutación de MCP con evolución fatal. Las recaídas con recuperación completa son bastante frecuentes y la progresión a IRT es menor que en la deficiencia de factor H. Dado que la MCP no es una proteína circulante, es menos probable el efecto beneficioso de las transfusiones de plasma o de las plasmaféresis, refiriéndose hasta un 70-100% de recuperación completa de los episodios agudos, independientemente de si se realiza plasmaféresis o no. La recurrencia en el riñón trasplantado no es frecuente, probablemente por la síntesis de esta proteína en el injerto38–40.
Factor IEl factor I, una proteasa de síntesis hepática, interviene en la regulación del complemento tanto de la vía clásica, como alternativa. Su función depende del factor H y de la MCP. El gen que codifica su síntesis se encuentra en el cromosoma 4q25, su mutación es causa de SHU atípico y puede coexistir con mutación del factor H. Puede manifestarse en los primeros meses de vida, aunque hay casos de inicio en adultos. Como en el déficit de factor H, el tratamiento incluye transfusiones de plasma y/o plasmaféresis. Aun así, la evolución a IRC es muy elevada, así como la recurrencia en el trasplante38–40,45.
Factor BLas mutaciones en el factor B del complemento incrementan la activación del complemento o retrasan su inactivación. Esto lleva a un déficit en la protección de las membranas celulares y es la alteración que se ha encontrado en varios miembros de 2 familias españolas con SHU atípico. Esta mutación presenta también una penetrancia incompleta, que en parte se explica por la necesidad de factores ambientales desencadenantes38,46.
Síndrome hemolítico urémico atípico asociado a deficiencia de la proteasa del factor de Von WillebrandEl factor de Von Willebrand es sintetizado por las células endoteliales y plaquetas e interviene en la adhesión y la agregación plaquetaria. Una metaloproteasa sintetizada en el hígado (ADAMTS-13) se encarga de su fraccionamiento en monómeros con menor capacidad agregante. El déficit de esta enzima favorece la formación de trombos en la microcirculación.
La deficiencia adquirida por autoanticuerpos contra esta enzima es la causa de la mayoría de las PTT encontradas en adultos y responde a plasmaféresis intensiva. En niños se han referido casos de SHU atípico en relación con deficiencia congénita de la proteasa del factor de Von Willebrand47. El tratamiento con transfusiones de plasma en estos casos puede revertir el proceso y prevenir las recaídas.
Síndrome hemolítico urémico asociado a alteraciones del metabolismo de la cobalaminaLos valores altos de homocisteína que se observan en la acidemia metilmalónica con homocisteinuria, secundaria a la alteración congénita en el metabolismo de la vitamina B12, probablemente son la causa de las manifestaciones vasculares. Se puede presentar en el período neonatal de forma fulminante o bien con un curso más insidioso en el niño mayor. La biopsia renal muestra microangiopatía trombótica38.
Lectura rápida
El pronóstico del SHU depende de la etiología. Los pacientes con SHU típico tienen mejor pronóstico, con mortalidad del 5%, con insuficiencia renal terminal del 5% y hasta el 25% con secuelas a largo plazo, como hipertensión arterial o algún grado de insuficiencia renal. Los pacientes con SHU secundario a infección por S. pneumoniae presentan un curso clínico más agresivo que los pacientes con SHU típico, más mortalidad (10–12%), especialmente los que presentan meningitis (38%); el 12% evoluciona a insuficiencia renal terminal, y el 16–25% de los casos permanece con enfermedad renal crónica o hipertensión arterial. El peor pronóstico se relaciona con los pacientes con alteración en el factor H del complemento, con un 60% de mortalidad e insuficiencia renal terminal el primer de evolución.
Descrito en pacientes adultos. Se produce por formación de anticuerpos dependientes de la quinina que reaccionarían con plaquetas, hematíes y células endoteliales38.
Síndrome hemolítico urémico asociado a cuadros clínicosLos cuadros clínicos asociados a SHU con etiología poco clara, referidos en la tabla 1, son poco frecuentes en niños, a excepción del SHU de los niños que se someten a trasplante de médula ósea. La etiología se desconoce, y puede estar en relación con el proceso en sí, o bien al acondicionamiento previo con fármacos, como ciclofosfamida, globulina antitimocito (ATG) o con radiación corporal total. La ciclosporina o el tacrolimus utilizados en el tratamiento de la enfermedad del injerto contra huésped también podrían ser la causa47.
Actitud diagnosticoterapéutica ante el paciente con síndrome hemolítico-urémicoLa importancia del enfoque diagnosticoetiológico correcto del paciente con SHU deriva de la necesidad de establecer de forma temprana pautas terapéuticas que pueden alterar la evolución de los pacientes con SHU atípico. Así, mientras en todos los pacientes es fundamental un tratamiento de soporte, evitar la administración de plasma en los pacientes con infección por S. pneumoniae28,29 o indicar de forma temprana la plasmaféresis37,49 en los pacientes con alteración del factor H puede ser determinante en su evolución.
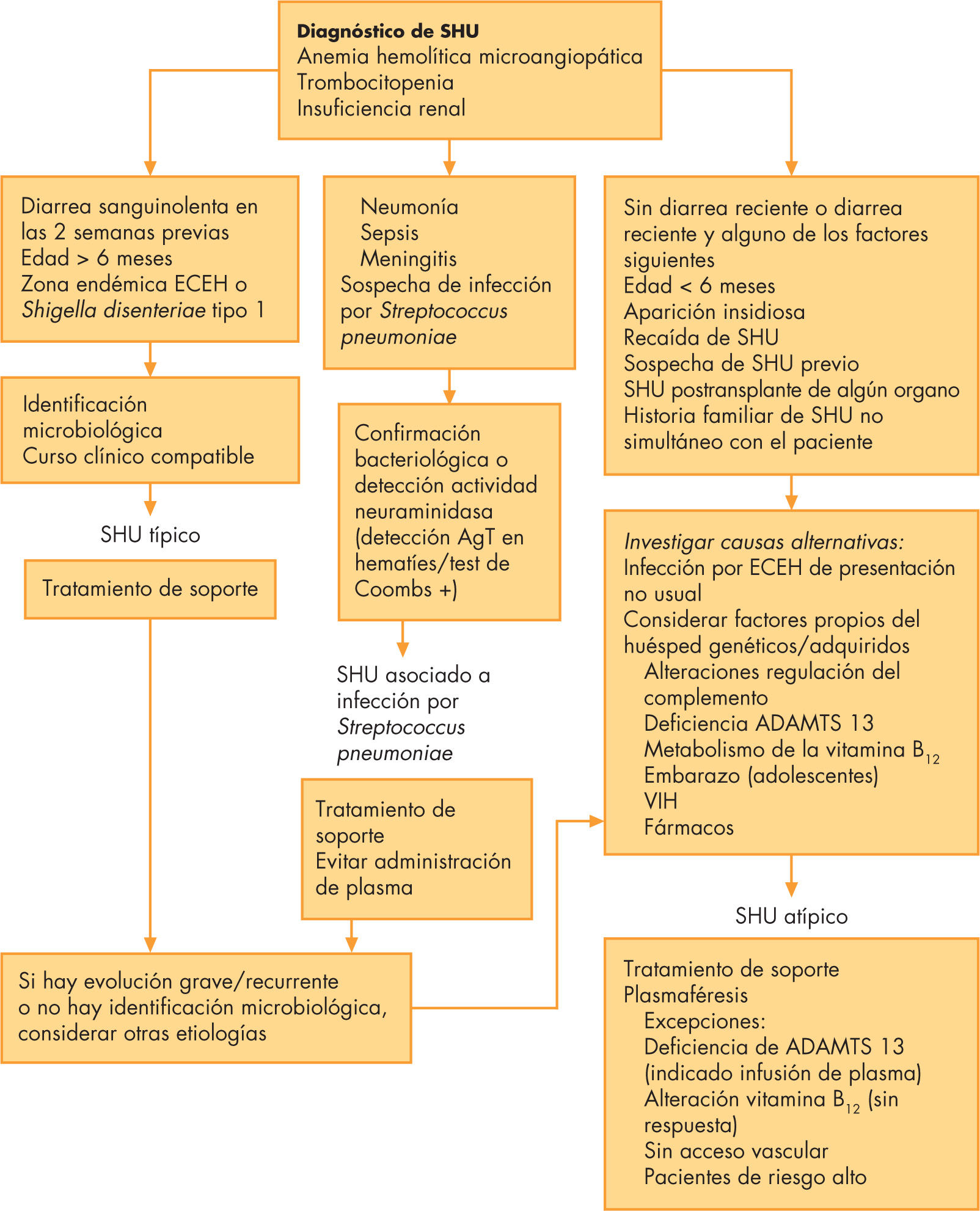
Por ello, se propone realizar un estudio exhaustivo de los pacientes con SHU de acuerdo a su presentación clínica, teniendo en cuenta que incluso en pacientes con causa infecciosa conocida puede haber otras causas genéticas o adquiridas que favorezcan la aparición del SHU49,50 (fig. 3).

Algoritmo diagnóstico terapéutico. ECEH: Escherichia coli enterohemorrágica; VIH: virus de la inmunodeficiencia humana; SHU: síndrome hemolítico urémico.