






Puntos clave
- •
El síndrome hemolítico urémico (SHU) se caracteriza por anemia hemolítica microangiopática, trombocitopenia e insuficiencia renal aguda.
- •
El SHU típico (90%) es más frecuente por el Escherichia coli 0157:H7. Mayor incidencia en menores de 2 años. Ocurre sobre todo en guarderías por contacto persona-persona. Principalmente, en verano y principios otoño. El pronóstico es favorable con curación en menos de 2 semanas.
- •
El SHU atípico (5%) debido a alteraciones del complemento puede aparecer al nacimiento y no hay diferencias estacionales. Tiene mal pronóstico con recurrencias frecuentes
- •
El SHU asociado a Streptococcus pneumoniae (5%), habitualmente con neumonía con empiema previo, es más grave que el típico con mayor mortalidad.
- •
El tratamiento es de soporte precisando diálisis con frecuencia. En la mayoría de formas asociadas a alteraciones del complemento es de elección la plasmaterapia.
- •
El eculizumab, anticuerpo recombinante anti-C5, está indicado en pacientes plasmarresistentes.
El síndrome hemolítico urémico (SHU) es un conjunto de signos y síntomas caracterizado por la presencia de anemia hemolítica microangiopática, trombopenia e insuficiencia renal aguda (IRA), constituyendo una de las causas más frecuente de IRA en la infancia con significativa morbimortalidad, por encima del 5% de mortalidad y un riesgo de insuficiencia renal crónica (IRC) o hipertensión arterial (HTA) del 25%1.
Descrito por Gasser en 1955, el SHU evoluciona desde formas autolimitadas hasta mortales. Clásicamente, se ha clasificado como típico o asociado a diarrea (D+ SHU) y atípico o no asociado a diarrea (D− SHU). Dentro del atípico, algunos autores incluyen el asociado a infección por neumococo (P− SHU). El SHU comparte características de la púrpura trombótica trombopénica (PTT), produciendo cuadros similares, incluso algunos autores hablan de una única entidad con diferentes presentación, más afectación renal en el SHU y mayor neurológica en la PTT2,3.
EtiologíaSíndrome hemolítico urémico asociado a diarreaEsta forma es la causa más frecuente de SHU en niños (90%) y ocurre con un episodio previo de diarrea habitualmente con sangre, relacionado con la Escherichia coli (E. coli) productora de la toxina Shiga (Stx) (STEC). La E. coli 0157:H7 es el serotipo más frecuentemente aislado. Un 2–15% de los pacientes infectados por esta cepa desarrollan un SHU. En mayo del 2011, el serotipo 0104:H4 causó un brote en Alemania con 3.816 infectados, 854 casos de SHU y 54 fallecimientos4.
Otra causa es la Shigella dysenteriae tipo 1, sobre todo en India, Bangladesh y Sudáfrica, con mayor mortalidad (15%) y evolución a IRC (40%)5. Otros cuadros infecciosos que desencadenan D+ SHU son Salmonella typhi, Campilobacter yeyuni, Yersinia, Pseudomonas, Bacterioides, Entaemoeba hystolitica y Aeromonas hydrofila.
El D+ SHU es más frecuente en < 2 años (6 meses-5 años) y tiene mayor incidencia en Argentina, Sudáfrica y Thailandia.
Sin diferencias de género, aunque en las mujeres las formas son más graves. Mayor incidencia durante el verano y el inicio del otoño. El principal lugar de contagio son las guarderías e instituciones cerradas, por la transmisión de persona a persona. La ingestión de carne es la causa de brotes de origen alimentario. También son frecuentes por productos contaminados por los animales, como lechuga, coles, repollo, etc.1.
Síndrome hemolítico urémico asociado a infección neumocócicaGeneralmente, ocurren sin diarrea. Debido a su mecanismo patogénico, la mayoría de los autores la consideran una entidad independiente del SHU atípico.
Es la forma más común de SHU sin diarrea y así, en un estudio realizado en 70 centros de Inglaterra, el 14% de los SHU entre 1998–2005 se asociaron a infección neumocócica6.
Aunque la infección neumocócica ha aumentado, el riesgo SHU es muy bajo (<1%).
Generalmente, asociada con procesos neumónicos y en el 51% con empiema7. Los serotipos identificados con mayor frecuencia son el 14, seguido del 6B y 23F8.
Síndrome hemolítico urémico atípico o no asociado a diarrea- 1.
Síndrome hemolítico urémico asociado a alteraciones del complemento. El 50% de los casos de D− SHU9 se debe a mutaciones en genes que regulan factores del complemento que incluye deficiencia o alteraciones del factor H, CD46 (antes proteína cofactor de membrana [MCP]), factor I, factor B, factor C3 y trombomodulina.
- 2.
Síndrome hemolítico urémico asociado a alteraciones de la proteasa del factor de Von Willebrand (ADMTS13). El factor de von Willebrand (FvW) interviene en la adhesión y agregación plaquetaria. La ADMTS13 sintetizada en el hígado escinde los largos multímeros, disminuyendo su capacidad agregante. Si hay déficit, se favorece la formación de trombos.
Está más relacionado con la PTT del adulto, aunque hay algunos casos de SHU en niños10.
- 3.
Síndrome hemolítico asociado a alteraciones del metabolismo de la cobalamina. El SHU puede complicar formas neonatales de aciduria metilmalónica con homocistinuria. El fallo de medro, el rechazo de las tomas, la hipotonía o la letargia pueden preceder al diagnóstico del cuadro. La mortalidad es extremadamente alta una vez desarrollado el SHU, debido a fallo multisistémico1,11.
- 4.
Existen otras causas de SHU de etiología conocida o desconocida, más frecuentes en adultos, incluidas en otra clasificación de consenso que algunos autores prefieren a la clásica3 (tabla 1).
Tabla 1.Clasificación etiológica de consenso el síndrome hemolítico urémico.
Categoría Características Nivel 1 Etiología conocida 1.1 Inducido por infección 1. Bacterias productoras de toxina Shiga y verocitotoxina como Escherichia coli enterohemorrágica, Shigella dysenteriae tipo1, Citrobacter 2. Streptococcus pneumoniae, neuraminidasa 1.2 Alteraciones en regulación del complemento 1. Genéticas 2. Adquiridas, como por ejemplo anticuerpos anti-factor H 1.3 Factor de von Willebrand, deficiencia ADAMTS 13 1. Genéticas 2. Adquiridas: autoinmunitaria, por fármacos 1.4 Deficiencia metabolismo cobalamina 1.5 Inducido por quinina Nivel 2 Asociación clínica: etiología desconocida 2.1 Virus de la inmunodeficiencia humana 2.2 Enfermedades malignas, quimioterapia y radiaciones ionizantes 2.3 Inhibidores de la calcineurina y trasplante 2.4 Embarazo, síndrome HELLP, anticonceptivo 2.5 Lupus eritematoso sistémico y síndrome antifosfolípido 2.6 Glomerulopatías 2.7 Familiar, no incluido en nivel 1 2.8 Inclasificado
Lectura rápida
El síndrome hemolítico urémico (SHU) se caracteriza por la presencia de manera simultánea de anemia hemolítica, trombopenia e insuficiencia renal aguda, siendo una de las causas más frecuente de insuficiencia renal aguda en la infancia.
Clásicamente se ha clasificado en SHU asociado a diarrea o típico (90%), debido fundamentalmente a Escherichia coli enterohemorrágicas productoras de toxina Shiga, y SHU atípico (5%), debido a alteraciones de factores de complemento, proteasas del factor de von Willebrand o alteraciones metabolismo de cobalamina y, dentro de este último grupo o de manera independiente, se incluye el SHU asociado a infecciones por Streptococcus pneumoniae (5%).
La patogenia del SHU asociado a diarrea se debe a la presencia de una toxina Shiga que finalmente provoca bloqueo de trascripción del ARN y síntesis proteica, condicionando este daño endotelial, la agregación plaquetaria y formación de microtrombos. Las formas asociadas a neumococo se deben a la neuraminidasa y las formas atípicas por alteración en la regulación de los factores del complemento y desencadenamiento de la cascada del mismo.
El SHU típico se presenta con pródromos de dolor abdominal, diarrea y, en ocasiones, vómitos, que preceden en 5–10 días al SHU. La clínica en cualquiera de las formas es similar con la presencia de anemia hemolítica microangiopática con esquistocitos, trombopenia, afectación renal en diferentes grados y pueden aparecer complicaciones a nivel neurológico, gastrointestinal, cardiovascular, pancreático o hepático.
El diagnóstico se realizará tras la sospecha clínica y con el apoyo de datos de laboratorio como la anemia hemolítica con bilirrubina directa elevada, haptoglobina disminuida y aumento significativo de LDH, trombopenia, alteración de urea y creatinina. El coprocultivo suele ser negativo cuando lleva evolución de más de 5 días. Es fundamental en las formas atípicas realizar un análisis del sistema complemento.
El pronóstico de las formas típicas suele ser favorable con resolución en 1–2 semanas. El SHU asociado a neumococo tiene peor pronóstico y en las formas atípicas este es variable, con recurrencias frecuentes dependiendo del factor alterado.
Son factores de mal pronóstico la presencia de leucocitosis, fiebre, oligoanuria persistente, severa colitis hemorrágica, afectación multisistémica grave y la histología renal muy alterada.
El tratamiento es fundamentalmente de soporte con fluidoterapia precoz antes de que el fallo renal se haya instaurado; el 50% de los pacientes requieren diálisis, siendo la técnica de elección dependiente del centro y del paciente.
Se debe ser restrictivo con las transfusiones de hematíes (hemoglobina menor de 6g/dl) y plaquetas (sangrado activo o para un procedimiento invasivo) para evitar sobrecarga hídrica.
La plasmaterapia como plasmaféresis o infusión de plasma fresco congelado, se ha demostrado eficaz en el SHU asociado a alteraciones de proteínas reguladoras del complemento salvo en alteraciones de la proteína cofactor de membrana (MCP) y se recomienda iniciar en las primeras 24 h del comienzo del cuadro.
Desde septiembre del 2011 se ha aprobado el tratamiento con eculizumab, un anticuerpo recombinante humano anti-C5. Se recomienda su uso en pacientes plasmarresistentes definidos por trombopenia y/o hemólisis y/o no mejoría de función renal tras 3–5 días de plasmaterapia. Disminuye la progresión a fracaso renal terminal y reduce las recurrencias postrasplante.
En los pacientes con fracaso renal terminal está indicado el trasplante renal, siendo el pronóstico más favorable en las formas asociadas a diarrea que en las del SHU atípico, donde tiene alto riesgo de recurrencias.
Se recomienda la plasmaterapia o el uso de fármacos bloqueadores como el eculizumab para la prevención de las complicaciones.
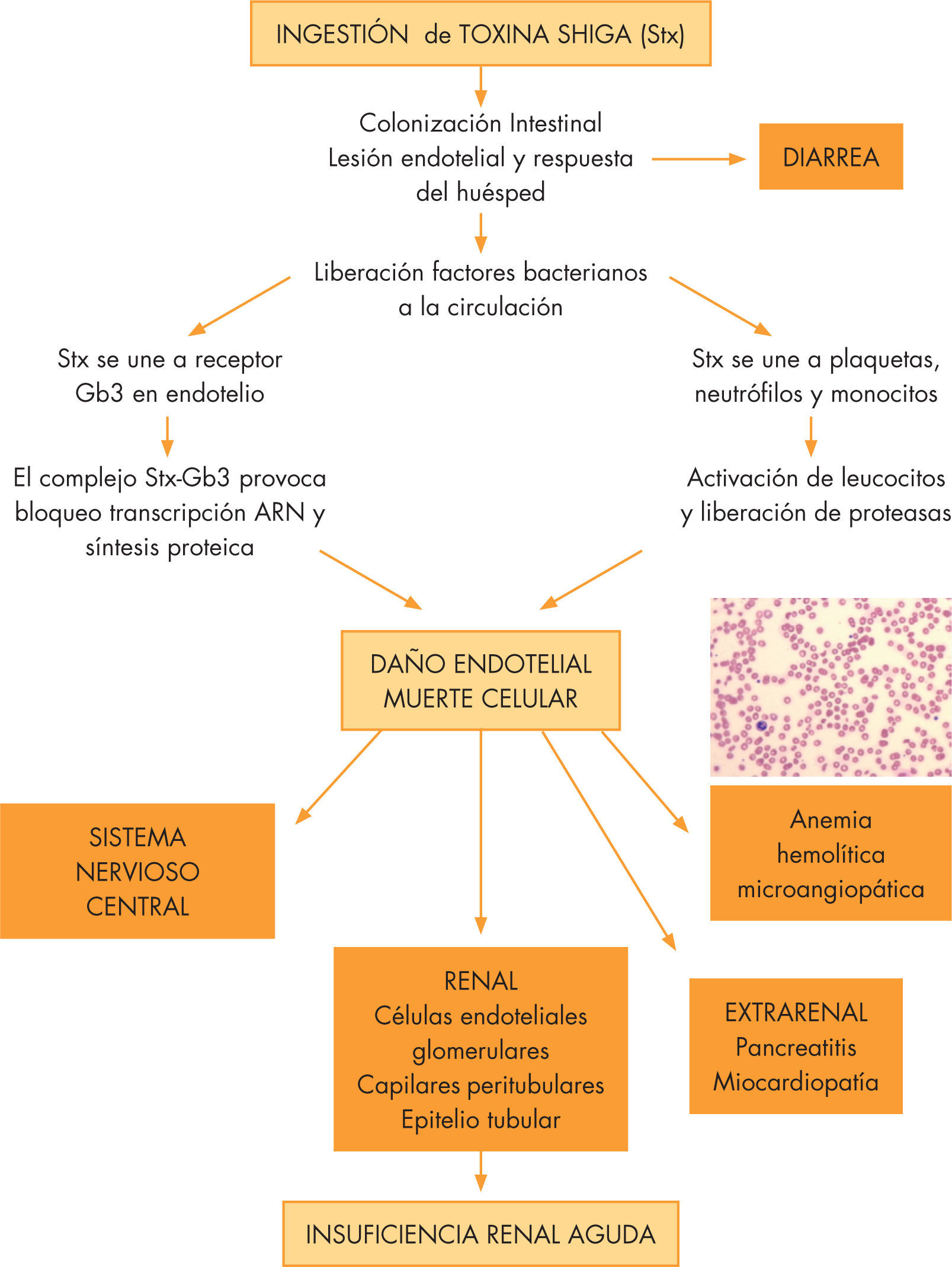
Está precedido por una diarrea causada por STEC (fig. 1). Tiene 3 factores de virulencia: la intimina, necesaria para la fijación a la pared intestinal facilitando el paso de la Stx a la circulación sistémica; la hemolisina, que inhibe el crecimiento de otras bacterias y provoca la hemólisis, y la Stx, que se une a un receptor (Gb3). La Stx presenta una subunidad A central y 5 subunidades B periféricas. Hay 2 tipos de toxina Shiga: Stx 1 y la Stx2. Se ha visto que las productoras de Stx2 se relacionan con cuadros más graves12. La cascada patogénica del D+ SHU se inicia con la ingestión del E. coli; la replicación bacteriana en el corion con la adhesión de la bacteria a la superficie celular, condicionando el paso de la toxina al torrente circulatorio, y se distribuyen a sitios ricos en receptor Gb3 presentes en riñón, cerebro, corazón, páncreas, pulmón y células (macrófagos, monocitos, células mesangiales y polimorfonucleares), uniéndose a través de la subunidad B de la Stx. La mayor expresión de Gb3 en el capilar glomerular comparado con otras células endoteliales puede explicar su especial preferencia por este órgano1,13. El complejo Stx-Gb3 se internaliza en el citoplasma y la subunidad A provoca el bloqueo de la trascripción del ARN y la síntesis proteica. La toxina Shiga estimula la producción por células endoteliales de citocinas como la interleucina 8. El daño en el endotelio favorece la agregación plaquetaria y el depósito de fibrina, apareciendo microtrombos con estrechamiento de la luz del vaso. A nivel renal, disminuye el filtrado glomerular y se desencadena la IRA. Al atravesar los estrechos capilares glomerulares, los hematíes son dañados y fragmentados, conduciendo a la anemia hemolítica microangiopática y la destrucción plaquetaria, a la que contribuyen anormalidades en los agentes plaquetarios (PgI2), agentes agregantes plaquetarios (Tx A2 y FvW)1,14.

No está definida la influencia de las alteraciones de la vía alternativa del complemento en la patogenia del D+ SHU. En diferentes estudios se ha visto en pacientes con D+ SHU alteraciones en los factores del complemento. Además, se ha visto que dichos factores se normalizaban a los 28 días del alta hospitalaria11,15.
Síndrome hemolítico urémico atípico asociado a alteraciones del complementoEl mayor progreso de la última década ha sido demostrar que la mayoría de los casos de SHU atípico se deben a alteraciones de la vía alternativa del complemento.
La activación se puede hacer por la vía clásica, vía de la lectinas o la vía alternativa, provocando finalmente la activación del factor C3. La vía alternativa del complemento está permanentemente activada como defensa frente a infecciones. Tienen una regulación estrecha para evitar que la célula endotelial sufra ataques por el depósito de C3b y la consecuente acción del complejo de ataque de membrana1,16 (fig. 2).

El sistema complemento mantiene un constante equilibrio con hidrólisis del C3 en el plasma. Esto produce la anafilotoxina C3a, la cual es proinflamatoria, y C3b, que se puede unir a la superficie de las células. Para prevenir que la cascada del complemento sea activada hay una serie factores reguladores, unos plasmáticos y otros celulares, que cooperan localmente para convertir el C3b en una molécula inactiva C3bi.
Entre los factores plasmáticos destacan el factor H, que inhibe la formación de C3 convertasa y acelera su destrucción, y el factor I, que interviene en vía clásica y alternativa13. Los factores en las células incluyen el CD46 o MCP, cuya alteración provoca daño endotelial por activación anómala del complemento y la trombomodulina. Mutaciones de los genes de estos factores se han visto en pacientes con SHU atípico con diferentes incidencias (tabla 2)1,13,17,18.
Características clínicas y pronóstico de pacientes con síndrome hemolítico urémico atípico asociado a alteraciones del complemento.
| Gen o subgrupo | Frecuencia de SHU | Mínima edad de comienzo | Riesgo de muerte o enfermedad renal terminal en primer episodio o antes de un año | Riesgo de recurrencias | Recurrencias después del trasplante renal |
| Factor H | 20-30% | Nacimiento | 50-70% | 50% | 75-90% |
| Factor I | 4-10% | Nacimiento | 50% | 10-30% | 45-80% |
| MCP | 5-15% | > 1 año | 0-6% | 70-90% | < 20% |
| C3 | 2-10% | 7 meses | 60% | 50% | 40-70% |
| Factor B | 1-4% | 1 mes | 50% | No en IRC | 100% |
| THBD | 3-5% | 6 meses | 50% | 30% | Un paciente |
| Anticuerpos antifactor H | 6% | 7-11 meses | 30-40% | 40-60% | Si altos anticuerpos presentes |
IRC: insuficiencia renal crónica; MCP: proteína cofactor de membrana; THBD: trombomodulina.
Más del 12% de las pacientes presentan varias combinaciones de mutaciones y entre el 6 y el 10% de los pacientes, sobre todo adolescentes, presentan anticuerpos frente al factor H.
Todos los defectos genéticos tienen como punto final aumentar la producción de C3 convertasa y secundariamente la liberación del complejo de ataque a la membrana en la superficie de la célula endotelial, fundamentalmente en la microcirculación del riñón17. Las células endoteliales se dañan y liberan radicales libres y proteinasas que aumentan el daño, incrementando adherencia plaquetaria y la formación de trombos en el riñón.
Síndrome hemolítico urémico asociado a infección neumococoEstá mediado por la toxina neuraminidasa1,19, que se une al acidosiálico de las glucoproteínas de la superficie de las células y expone el Ag de Thomsen Friedenreich (Ag T) en hematíes, plaquetas y glomérulo. En el plasma, hay inmunoglobulinas anti-T que reaccionan produciendo daño en el hematíe y en el riñón20. Esto explicaría la positividad del test de Coombs directo, a diferencia de otras formas de SHU.
No se han descrito alteraciones genéticas en la regulación del complemento en estas formas de SHU, por lo que no existen recurrencias.
ClínicaEl período de incubación entre la infección por E. coli y los síntomas es entre 3–5 días21. Presenta una fase prodrómica con dolor abdominal, en ocasiones vómitos y diarrea (80% con sangre) que preceden al SHU en 5–10 días (fig. 3).

El riesgo SHU se incrementa si el paciente es muy joven, ha recibido fármacos antimotilidad intestinal o antibióticos22.
Las características clínicas del SHU sea cual sea su origen incluyen:
- 1.
Hematológico. La anemia hemolítica ocurre en todos los pacientes con SHU. Se caracteriza por1:
- –
Hemoglobina menor de 8g/dl.
- –
Test de Coombs directo negativo salvo en las formas asociadas a neumococo (90%).
- –
Frotis de sangre periférico con esquitocitos (> 10%).
La hemólisis produce elevación de la bilirrubina indirecta y disminución de haptoglobina. La lactatodeshidrogenasa (LDH) está muy elevada23.
Las plaquetas están por debajo de 40.000/mm3. No suele haber púrpura ni sangrado evidente. No hay correlación entre la plaquetopenia y la anemia con la afectación renal.
- –
- 2.
Riñón. La afectación renal va desde cuadros de hematuria y proteinuria a severa afectación renal con oligoanuria (50% afectación renal grave). La mayoría presentan hematuria microscópica2.
La oligoanuria ocurre en un 50–60% de los pacientes, con una duración media de una semana. Un 60% de los pacientes requieren diálisis durante 10 días24.
- 3.
Sistema nervioso central (SNC): La afectación del SNC puede deberse a isquemia cerebral por microtrombos, por efecto de la HTA, por hiponatremia o por asociación de las anteriores. Más frecuente en el D− SHU, aunque pueden aparecer hasta en un 20% de las asociadas a diarrea2,24.
La clínica incluye convulsiones, coma, accidente cerebrovascular, hemiparesia, ceguera cortical y otras focalidades neurológicas25.
- 4.
Gastrointestinal. Se puede afectar cualquier área desde el esófago al recto. Las complicaciones incluyen perforación con necrosis intestinal, prolapso rectal y peritonitis26.
- 5.
Cardiovascular. Puede producirse insuficiencia cardiaca por isquemia miocárdica. La HTA es frecuente. Ambas complicaciones se agravan por la sobrecarga de volumen2,22,27.
- 6.
Páncreas. La afectación pancreática leve es muy frecuente y en raras ocasiones es grave con necrosis, seudoquistes o ambos. Puede provocar una diabetes insulinodependiente que generalmente es transitoria. Hasta el 10% desarrolla intolerancia a la glucosa.
- 7.
Hígado. Es frecuente la hepatomegalia que puede ir acompañada de elevación de las transaminasas.
Un paciente con historia de diarrea, sobre todo con sangre, que presenta alteración multisistémica requiere una evaluación precisa para detectar el posible SHU.
LaboratorioAparte de los hallazgos de anemia hemolítica y plaquetopenia, podemos encontrar leucocitosis con desviación a la izquierda que puede llegar a 50–60.000/mm3. La coagulación suele estar normal.
La urea y la creatinina estarán elevadas, con alteraciones electrolíticas en relación con los vómitos, diarrea o IRA. Puede existir hiperuricemia por la insuficiencia renal, la deshidratación y la rotura celular.
Estudio de las hecesLa bacteria E. coli está presente en las heces durante unos pocos días, por lo que el rendimiento del coprocultivo es bajo tras 7 días de diarrea. STEC no crece en cultivos rutinarios y se usan placas de agar McConkey. El diagnóstico del SHU es clínico y no puede descartarse por un coprocultivo negativo.
La toxina Stx puede ser detectada usando test específicos, estudios genéticos o mediante técnica de ELISA.
Estudio del complementoConsiderando las implicaciones pronósticas y terapéuticas, se debería realizar un análisis del complemento y un estudio genético de los genes codificadores16.
Estos estudios están limitados a un número escaso de laboratorios y son muy caros y de uso exclusivo en investigación.
Un consenso del Grupo de Trabajo Europeo sobre Genética ha descrito recientemente un protocolo para la detección de mutaciones en el SHU atípico28.
PronósticoLas manifestaciones hematológicas del D+ SHU suelen resolverse en 1–2 semanas. El pronóstico renal suele ser favorable, resolviéndose después de la mejoría hematológica. La mortalidad es menor del 5%, pero otro 5% de los pacientes tiene importantes secuelas. Resultados similares se han visto durante el brote de Alemania en el año 2011 por E. coli 0104:H429. Indicadores asociados a peor pronóstico son:
- –
Leucocitosis, frecuente en el SHU típico, > 20.000/mm3. La fiebre (5–20%) empeora el pronóstico23.
- –
Oligoanuria inicial que es persistente (> 5 días de anuria o > 10 días de oliguria)30.
- –
Severa colitis hemorrágica con prolapso rectal o isquemia colónica.
- –
Afectación multisistémica importante.
- –
Persistente proteinuria.
- –
Histología renal mostrando microangiopatía glomerular con afectación mayor del 50% de glomérulos, microangiopatía arterial y/o necrosis cortical.
El pronóstico a largo plazo para los supervivientes de las formas de D+ SHU es desconocido. Un estudio con seguimiento durante 5 años demostró cifras de presión arterial normal y niveles de creatinina discretamente más elevados en los pacientes que presentaron un SHU frente al grupo control31.
El SHU asociado a neumococo presenta peor pronóstico que el SHU típico32. Aproximadamente, un 16% de estos pacientes mantienen HTA e IRC, y el 10% evoluciona hacia formas terminales de insuficiencia renal33,34.
Las formas atípicas de SHU asociado a alteraciones del complemento se caracterizan por recaídas frecuentes, alta mortalidad y evolución a IRC muy frecuente17,18,35 (tabla 2).
La creatinina durante el primer episodio es el único factor pronóstico en cuanto a evolución a IRC terminal o fallecimiento en el primer año de vida36.
TratamientoEl tratamiento del SHU es fundamentalmente de soporte con algunas consideraciones en las formas atípicas.
FluidoterapiaDebe ser precoz ya que está relacionado con menor riesgo de oligoanuria en D+ SHU37. En un estudio en 29 niños con D+ SHU que evolucionaron a oligoanuria y necesitaron diálisis, estos habían recibido menos líquido durante los primeros 4 días del cuadro diarreico38. Con oligoanuria, la reposición hídrica debe ser limitada (pérdidas insensibles y diuresis).
Tratamiento del fallo renal agudoAproximadamente, el 50% de los pacientes con D+ SHU requieren diálisis precoz si el paciente presenta sobrecarga hídrica, hiperpotasemia, acidosis, hiponatremia u oligoanuria sin respuesta a diuréticos.
La elección de la técnica depende de la experiencia del centro y del paciente:
- –
La diálisis peritoneal es muy utilizada en pediatría por ser técnicamente sencilla y bien tolerada sobre todo en niños pequeños
- –
La hemodiálisis es adecuada en pediatría y es preferible si el niño presenta dolor abdominal con mala tolerancia a los pases de diálisis peritoneal. Las técnicas continuas de reemplazo renal son preferibles en situaciones de inestabilidad hemodinámica y cuando necesiten un estricto control del volumen1.
La diálisis no cambia el curso de la enfermedad, su uso preventivo no está indicado. Los pacientes requieren habitualmente entre 5–7 días de terapia.
Tratamiento de las alteraciones hematológicasHasta el 80% de los niños con SHU requieren transfusión de hematíes. Se debe ser restrictivo y trasfundir solo cuando la hemoglobina sea menor de 6g/dl, manteniendo niveles entre 8–9g/dl, evitando la sobrecarga de volumen y mantener una baja viscosidad de la sangre.
Se recomienda, en las formas asociadas a neumococo, transfundir con hematíes para que no contengan IgM y aumenten la reacción Ag-Ac.
Transfundir plaquetas si existe un sangrado activo o para un procedimiento invasivo39. Es poco frecuente que se tenga que transfundir porque no suelen disminuir menos de 10.000/mm3. En un estudio retrospectivo40 no hubo sangrado en ninguno de los 73 pacientes que requirieron algún procedimiento invasivo.
Tratamiento de complicaciones- –
La HTA se trata evitando la sobrecarga de volumen y con fármacos antihipertensivos. Los antagonistas del calcio como amlodipino o isradipino son de elección en pediatría.
Los inhibidores de la enzima conversiva de angiotensina (IECA) se utilizan para las secuelas a largo plazo porque además parece que disminuyen la excreción de proteínas, lo que puede retardar la evolución hacia enfermedad renal crónica.
- –
Si el dolor abdominal es muy intenso, se deben evitar los AINE por su nefrotoxicidad. El opioide de elección sería el fentanilo, porque no tiene metabolitos activos.
- –
No está indicado el uso de antibióticos porque no previenen la evolución a SHU22.
- –
El uso de plasmaterapia, recomendado para pacientes con SHU atípico, no ha demostrado su eficacia en el D+ SHU. Algunos centros lo han utilizado como terapia de rescate, sobre todo en afectaciones neurológicas graves41.
- –
La anticoagulación con heparina, urocinasa o dipiridamol no ha conseguido mejorar el pronóstico42.
- –
El uso de corticoides tampoco ha demostrado mejoría.
- –
Se ha intentado bloquear la toxina Stx con diferentes fármacos, sin resultados43.
- –
Se están estudiando anticuerpos monoclonales específicos frente la toxina Stx1,44.
- 1.
Plasmaféresis. La plasmaféresis es la terapia de elección en SHU atípico asociado a alteraciones del complemento. Diferentes estudios han demostrado una disminución de la mortalidad del 50 al 25% desde su uso17,45.
Con la plasmaféresis se eliminan los factores del complemento alterados, así como otros factores estimuladores del daño endotelial y de la hiperagregación plaquetaria, reponiéndolos con plasma fresco congelado. El tratamiento intensivo con plasmaféresis puede prevenir recurrencias y evolución a estadios terminales de IRC.
El 63% de los pacientes con mutaciones del factor H que recibieron plasmaterapia tuvieron respuesta, pero solo el 5% una recuperación completa; en mutaciones del factor I, solo el 25% respondió. En alteraciones del MCP, al no ser un factor circulante, no está indicada la terapia, evolucionando a resolución en el 90%. Los beneficios de la plasmaterapia en pacientes con mutaciones en C3, factor B o trombomodulina no están claramente definidos, aunque se han visto repuestas. La plasmaféresis es indicación absoluta en pacientes con anticuerpos anti-factor H.
Las guías actuales recomiendan iniciar la terapia en primeras 24 h de inicio del cuadro, aún sin resultados del complemento46,47. Se recomienda un intercambio de 1,5 veces el volumen de plasma y reemplazarlo con plasma fresco congelado o crioprecipitado. Continuar hasta un adecuado control de la enfermedad:
- –
Plaquetas > 150.000/mm3.
- –
Cese de la hemólisis con normalización de LDH.
- –
Mejoría de la función renal.
Si la plasmaféresis inicial es efectiva, se realizarán 5 sesiones cada semana durante 2 semanas, seguidas por 3 sesiones por semana durante las siguientes 2 semanas. Una vez controlada la enfermedad, la frecuencia en el siguiente mes no está establecida y depende de factores individuales del paciente. En general, la plasmaféresis suele ser suspendida antes de la indicación para ello, por problemas técnicos, y aunque algunos evolucionan bien, otros progresan a fracaso renal terminal o recurren1,45.
- –
- 2.
Infusión de plasma. Cuando la plasmaféresis no está disponible o no puede ser aplicada inmediatamente, se recomienda la administración de plasma fresco congelado. Con esto se administran los factores ausentes o anormales del complemento o fvW. Las ventajas es que no se requiere ningún equipo específico ni vía central. Generalmente se infunden 20–30ml/kg de peso. La frecuencia de administración y la respuesta son similares a las de la plasmaféresis1,17.
Los problemas de esta terapia son la sobrecarga de volumen y la hiperproteinemia, que se observa en la administración crónica del plasma.
Bloqueadores del complemento: eculizumabEl eculizumab es un anticuerpo recombinante humano anti-C5, que bloquea el paso de C5 a C5b y, por tanto, la generación del MCP (C5b-C9) (fig. 2)1,48.
Es el primer tratamiento aprobado por la Food and Drug Administration (2011) y posteriormente por la Agencia Europea del Medicamento para adultos y niños con SHU atípico. La mayoría de las guías recomiendan su uso en plasmarresistentes definidos por persistente trombocitopenia y/o hemólisis, y/o no mejoría de la función renal tras 3–5 días de plasmaterapia diaria. Algunos autores lo consideran incluso como un fármaco de primera línea en caso de diagnostico inequívoco de SHU atípico48.
Los estudios realizados confirman que la administración de eculizumab es efectiva para el proceso de microangiopatía angiopática, aumentando las plaquetas, cesando la hemólisis y estabilizando la función renal1,48,49.
Ha sido utilizado para prevenir y tratar las recurrencias postrasplante, siendo mejor estrategia utilizarlo de manera preventiva.
Se ha usado incluso en menores de un mes y en pacientes con D+ SHU con afectación neurológica importante con buenos resultados49,50.
El coste del fármaco obliga a definir claramente sus indicaciones. La dosis en niños debe ser ajustada según el peso, aunque hay poca experiencia, la recomendación es dosificar según bloqueo del complemento (CH50). Dada la inhibición del complemento, así como la alteración de la función de neutrófilos y monocitos que produce el fármaco, estos pacientes son susceptibles a infecciones por bacterias encapsuladas, por lo que deben ser vacunados frente a Neisseria meningitidis, Streptococcus pneumoniae y Haemophilus influenzae.
Trasplante renalLos pacientes con permanente fallo renal debido a D+ SHU tienen riesgo de recurrencias bajo y se puede realizar el trasplante renal como otras enfermedades renales.
El trasplante renal en pacientes con SHU atípico es más dificultoso por el alto riesgo de recurrencias y rechazo, muy dependiente del tipo de alteración que presente (tabla 2). El trasplante hepático aislado o hepático-renal puede ser curativo, sobre todo en alteraciones del factor H. La mayoría de los autores recomiendan la plasmaterapia o el uso de fármacos bloqueadores, como el eculizumab, para la prevención de las complicaciones1.
Perspectivas de futuroLos próximos años deben servir para aclarar las dudas sobre la implicación del complemento en el D+ SHU, avanzar en el desarrollo de nuevos agentes, como anticuerpos monoclonales o moléculas recombinantes que neutralicen la Stx o bloqueen el complemento, así como para definir las pautas y la duración, indicaciones tanto de plasmaterapia como con eculizumab, estableciendo las indicaciones del trasplante hepático y/o renal como curación definitiva de la enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.