



Puntos clave
- •
El diagnóstico y el tratamiento de las diferentes formas clínicas de linfohistiocitosis hemofagocítica (HLH) requiere un equipo multidisciplinar.
- •
La citotoxicidad de las células natural killer (NK) y la citometría de flujo constituyen 2 herramientas diagnósticas imprescindibles para el diagnóstico de HLH.
- •
Existen mutaciones desconocidas, otros genes implicados y alteraciones epigenéticas que podrían explicar las presentaciones atípicas y las recurrencias de muchas formas secundarias. Por lo que la ausencia de un diagnóstico genético no excluye las formas primarias.
- •
El tratamiento, según el protocolo HLH-2004, debe instaurarse en todas las formas primarias y en las secundarias con presentación grave o recurrente, incluso con citopenias intensas y disfunción multiorgánica.
- •
Las formas refractarias pueden tratarse con protocolos de segunda línea que incluyan globulina antitimocito, alentuzumab, etanercept o vincristina.
- •
El trasplante de progenitores hematopoyéticos con acondicionamiento de intensidad reducida constituye el tratamiento de elección para las formas primarias y las secundarias refractarias o con persistencia de la actividad citotóxica NK disminuida.
El diagnóstico de linfohistiocitosis hemofagocítica (HLH) requiere confirmación genética y/o presentar al menos 5 criterios clínicos: fiebre, esplenomegalia, bicitopenia, hipofibrinogenemia o hipertrigliceridemia, hiperferritinemia, elevación del receptor soluble de la interleucina 2 (IL-2), disminución o ausencia de la actividad citotóxica de las células natural killer (NK) y la presencia de hemofagocitosis en la médula ósea, el bazo o los ganglios linfáticos1. En ausencia de tratamiento adecuado, la HLH presenta un curso fulminante en pocos meses. El tratamiento con etopósido (VP16) ha permitido alcanzar remisiones a corto plazo del 90%. Sin embargo, el origen genético de la enfermedad y las reactivaciones hacen necesario el trasplante de progenitores hematopoyéticos (TPH), estabilizando la supervivencia a largo plazo alrededor del 60% y hasta el 80% con acondicionamientos de intensidad reducida2,3. Los defectos genéticos constituyen la diana para el desarrollo de la terapia génica a medio corto plazo.
DiagnósticoEs fundamental realizar el diagnóstico precoz para así indicar el tratamiento adecuado. La ausencia de síntomas, signos o biomarcadores patognomónicos requiere un elevado índice de sospecha. La Sociedad Internacional del Histiocito propuso en su última revisión los criterios diagnósticos de HLH (tabla 1)1. Una de las principales limitaciones de estos criterios es que no diferencian las formas primarias de las secundarias. La presentación en edades muy tempranas, la afectación neurológica, la trombopenia intensa y una desmesurada elevación de la ferritina (> 10.000mg/l) y del receptor soluble de la IL-2 (sCD25) son indicativas de formas primarias4–6.
Criterios diagnósticos revisados de linfohistiocitosis hemofagocítica (HLH) según el protocolo internacional HLH 2004.
| El diagnóstico de HLH puede establecerse si se cumple uno de los 2 criterios siguientes |
| 1. Diagnóstico molecular consistente con HLH |
| 2. En ausencia de diagnóstico molecular, cuando se cumplan 5 de los siguientes criterios: |
| 1. Fiebre |
| 2. Esplenomegalia |
| 3. Citopenias en sangre periférica que afecten al menos a 2 de los 3 linajes: |
| Hemoglobina < 9g/dl (en neonatos <10g/dl) |
| Plaquetas < 100 × 109/L |
| Neutrófilos < 1 × 109/l |
| 4. Hipertrigliceridemia (> 265mg/dl) y/o hipofibrinogenemia (fibrinógeno < 150mg/dl) |
| 5. Hemofagocitosis en médula ósea, ganglio o bazo |
| Criterios adicionales |
| 6. Disminución o ausencia de actividad citotóxica de las células natural killer |
| 7. Hiperferritinemia (> 500ng/ml) |
| 8. Elevación del receptor soluble de la interleucina 2 (sIL-2R) > 2.400U/ml |
| Comentarios adicionales |
| 9. Otros síntomas clínicos y hallazgos de laboratorio que nos sugieren que estamos ante una linfohistiocitosis hemofagocítica familiar son: síntomas neurológicos, adenopatías, ictericia, rash cutáneo, hepatopatía, hipoproteinemia, hiponatremia, elevación de las lipoproteínas de muy baja densidad del colesterol, disminución de las proteínas de alta densidad del colesterol, en el líquido cefalorraquídeo, pleocitosis y/o hiperproteinorráquea |
| 10. En absoluto, la ausencia de hemofagocitosis en médula ósea no excluye el diagnóstico. Valorar realizar aspirados de médula ósea seriados o buscarla en otras localizaciones (adenopatías, bazo, hígado) |
| 11. Hiperferritinemia > 10.000ng/ml son muy indicativas de HLH |
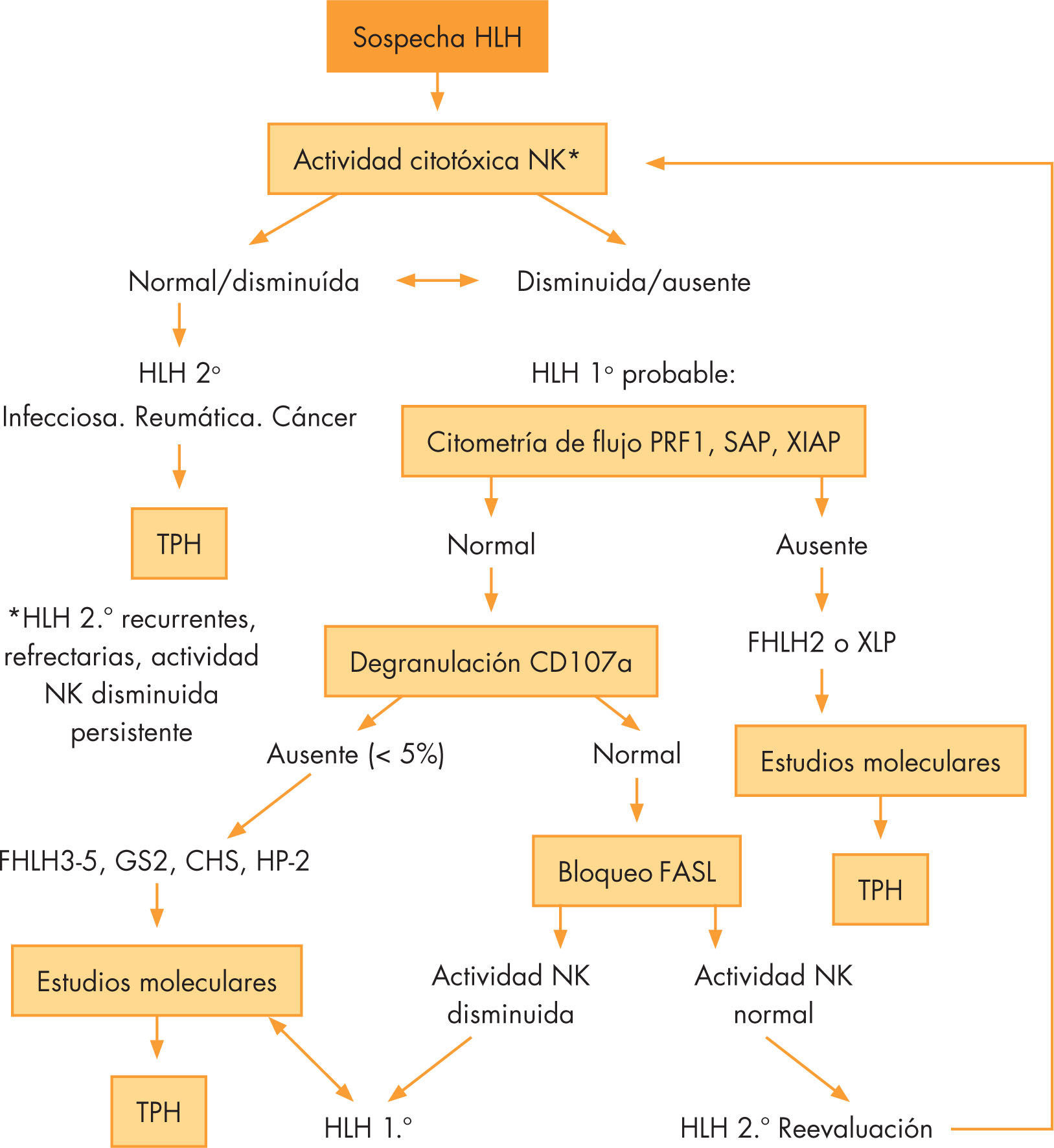
Atendiendo a su fisiopatología, la HLH tiene en común un deterioro de la citotoxicidad celular mediada por los linfocitos T citotóxicos (CTL) y las células NK7. Una actividad citotóxica disminuida es muy indicativa de HLH primaria, mientras que la normalidad es más probable en las formas secundarias. En las formas primarias esta alteración está presente antes de que aparezcan los síntomas y no se recupera tras el tratamiento. La citometría de flujo permite determinar las proteínas PRF1, SAP y XIAP en las células NK y CTL. La ausencia de estas proteínas indica una forma primaria familiar, FHLH-2, o el síndrome linfoproliferativo ligado al X, XLP1 y XLP2, respectivamente. Por el contrario, la presencia de estas proteínas excluye estos diagnósticos. En estos casos, debemos determinar la expresión en la membrana celular de NK y CTL de la proteínas lisosomal CD107a. Esta proteína determina la capacidad de exocitosis de los gránulos de perforina-granzima de las células efectoras. Una degranulación < 5% es muy indicativa de HLH primaria con defectos en la degranulación como las formas familiares, FHLH-3, 4 y 5, y las inmunodeficiencias como el síndrome de Griscelli tipo II y el síndrome de Chédiak Higashi8.
Diagnóstico funcional: actividad citotóxica natural killerPese a constituir un hecho determinante en la fisiopatología de la HLH, la actividad citotóxica de las células NK, además de no estar accesible para la mayoría de los centros sanitarios, tiene limitaciones para su interpretación diagnóstica: a) la presencia de falsos negativos cuando existe linfopenia; b) falsos positivos en el pacientes crítico con activación de mecanismos citotóxicos compensadores, y c) no informar sobre el mecanismo subyacente a la alteración funcional.
Lectura rápida
El diagnóstico de la linfohistiocitosis hemofagocítica (HLH) es complejo. Requiere de un alto grado de sospecha clínica y la combinación de datos clínicos y de laboratorio, incluyendo variables inmunológicas. La presencia de hemofagocitosis en médula ósea, ganglio linfático o hígado no es en absoluto necesaria para realizar el diagnóstico.
La actividad citotóxica de las células natural killer (NK) constituye, con limitaciones, una prueba muy importante para orientar el diagnóstico de HLH. La disminución de la actividad citotóxica NK se describe en prácticamente todas las formas primarias con presentación clínica grave y no se recupera tras el tratamiento. Sin embargo, las formas primarias con presentación atípica y la mayoría de las formas secundarias presentan menor variabilidad de la actividad NK, además de normalizarse cuando desaparecen los síntomas clínicos.
La citometría de flujo constituye una herramienta diagnóstica muy importante para el diagnóstico de las formas primarias de HLH, ya que permite identificar las proteínas perforina, SAP y XIAP, orientando al diagnóstico de FHLH2, XLP1 y XLP2 respectivamente. Adicionalmente, los ensayos de degranulación por citometría de flujo mediante la determinación de la glucoproteína CD107a permite identificar las formas familiares FHLH-3, 4 y 5 y las inmunodeficiencias GS2, CHS, H-P2.
Nuevas herramientas diagnósticas se están desarrollando en el momento actual como la determinación sinérgica en suero de las proteínas solubles sCD25 y sCD63, y estudios de degranulación por citometría de flujo en subpoblaciones de linfocitos específicas, CD57+.
Las pruebas genéticas deben incluir el análisis de mutaciones en los genes PRF1, UNC13D, STX11, STXBP2, SH2D1A y BIRC4. Sin embargo, en la mitad de los pacientes con HLH desconocemos el defecto genético. En estos casos sospecharemos alteración genética subyacente cuando el cuadro clínico sea grave, refractario y recurrente, exista asociación familiar y persista defecto permanente en la citotoxicidad NK.
La sepsis, el síndrome de respuesta inflamatoria sistémica y la disfunción multiorgánica en muchas ocasiones cumplen criterios diagnósticos de HLH, indicando un proceso fisiopatológico relacionado que podría beneficiarse del tratamiento con fármacos antiinflamatorios e inmunosupresores.
El tratamiento debe iniciarse sin demora ante la sospecha clínica, especialmente en las presentaciones graves con disfunción neurológica y/o pulmonar. La precocidad del tratamiento adecuado es clave para la supervivencia del paciente.
Las bases del tratamiento están dirigidas a detener la inflamación mediante: a) fármacos antiinflamatorios con buena penetración en el sistema nervioso central (dexametasona), y b) fármacos inductores de apoptosis en las células responsables de la inflamación (linfocitos T citotóxicos, NK y macrófagos) mediante quimioterápicos potentes como los inhibidores de epidofilotoxinas (etopósido)
Las formas secundarias deben manejarse cuidadosamente, reservando el tratamiento según protocolo HLH-2004, para las formas graves, recurrentes o que no mejoran con el tratamiento etiológico.
El trasplante alogénico de progenitores hematopoyéticos (TPH) es actualmente el único tratamiento curativo para las formas primarias y para algunas causas secundarias muy graves o recurrentes. Los mejores resultados del TPH se alcanzan en pacientes en remisión estable y sin actividad de la enfermedad. Sin embargo, la ausencia de respuesta no debe ser obstáculo para el TPH en las formas primarias. Los nuevos acondicionamientos menos intensos han disminuido las complicaciones derivadas del acondicionamiento mieloablativo, permitiendo situaciones de quimerismo mixto suficientes para evitar los episodios de HLH.
Los objetivos futuros incluyen descubrir nuevas mutaciones en genes conocidos, nuevas alteraciones genéticas, desarrollar herramientas diagnósticas, algoritmos de estratificación y tratamientos de rescate eficaces y poco tóxicos. En este sentido, en el momento actual se está desarrollando ensayos clínicos con inhibidores de citocinas como el interferón.
La terapia génica en las formas genéticas conocidas podría constituir a largo plazo un tratamiento curativo que reemplace al TPH; sin embargo, en el momento actual se encuentra en estadios de desarrollo muy preliminares.
La disminución de la actividad citotóxica NK se ha observado tanto en formas primarias como secundarias de HLH, sobre todo en fase aguda, mejorando en las formas secundarias tras la desaparición de los síntomas y permaneciendo en las formas primarias. Se han descrito diferentes tipos de alteración de la actividad citotóxica NK: tipo I, déficit cualitativo de la actividad citotóxica sensible a la estimulación con IL-2 y con recuentos normales; tipo II, déficit cualitativo insensible a la estimulación con IL-2 y con cifras normales de células NK, y tipo 3, déficit cualitativo de las células NK insensible a la estimulación con citocinas y que asocia déficit cuantitativo de esta población.
La conservación de la actividad citotóxica de las células NK tampoco descarta HLH, ya que la variabilidad genotípica de las formas primarias puede inducir fenotipos de actividad NK conservada.
A pesar de estas limitaciones, la disfunción de las células NK ha demostrado ser un ensayo muy consistente, fácil de realizar, complementario a otras pruebas y adecuado para fines diagnósticos9.
Diagnóstico fenotípico: citometría de flujoLa citometría de flujo permite analizar diferentes proteínas específicas de las HLH primarias en diferentes subpoblaciones celulares. La principal limitación de esta prueba diagnóstica la constituye las citopenias profundas, por lo que el estudio de citometría de flujo puede posponerse tras el tratamiento y mejoría de los síntomas. Sin embargo, la presencia de la proteína no garantiza su funcionalidad y su ausencia ocurrir en presencia del gen, manifestando diferentes alteraciones implicadas en la regulación génica, epigenética, la transcripción y/o la estabilidad del ARN mensajero. Por lo tanto, las pruebas funcionales complementan las pruebas genéticas y fenotípicas. Las mutaciones en el gen PRF1 son la causa más común de FHLH. La marcación intracelular de la proteína perforina permite determinar su expresión en diversas poblaciones celulares. Defectos genéticos en homocigosis en el gen PRF1 asocian una marcada disminución o ausencia de la expresión de perforina. La ausencia completa es generalmente causada por la introducción de un codón de parada prematuro o una mutación en el gen, que origina una proteína truncada. Mutaciones de sentido erróneo pueden también causar reducciones en la expresión de perforina. En los casos en que exista sospecha clínica de XLP, el análisis por citometría de flujo de las proteínas SAP y/o XIAP puede ayudar al diagnóstico. La expresión de SAP y XIAP es mutuamente excluyente. El déficit en SAP constituye el 55–60% de todos los XLP, por lo que XIAP se testará en los casos en los que la expresión de SAP sea normal10,11.
Diagnóstico funcional por citometría de flujo: ensayos de degranulaciónLa glucoproteína asociada a la membrana lisosomal (LAMP-1), CD107a, se trasloca a la membrana de las células CTL y NK tras estimulación y muestra una fuerte correlación con la producción de citocinas y la actividad citotóxica. Diferentes ensayos de experimentación que comparan la expresión basal y tras estimulación han demostrado deficiencias en MUNC13-4 y la sintaxina-11. Por lo tanto, la translocación de CD107a a la superficie de la célula efectora se presenta en pacientes con defecto en el transporte vesicular debido a mutaciones en homocigosis en UNC13D, STX11. Similar a este ensayo, la transferencia de CD63 (LAMP-3) de la membrana de los gránulos a la membrana celular es un indicador de degranulación y citotoxicidad11.
Una adecuada interpretación de estas pruebas diagnósticas (figs. 1 y 2), y en espera de las pruebas genéticas, puede ayudarnos a orientar de manera adecuada el tratamiento y el consejo genético.

Algoritmo diagnóstico de HLH según su fisiopatología.
FASL: FAS ligando; FHLH: linfohistiocitosis hemofagocítica familiar; HLH: síndromes hemofagocíticos; NK: natural killer; SCH: síndrome de Chédiak Higashi tipo II; SGII: síndrome de Griscelli tipo II; SHPII: síndrome de Hermansky-Pudlak tipo II; TPH: trasplante de progenitores hematopoyético; XSLP: síndromes linfoproliferativos ligados al cromosoma X.
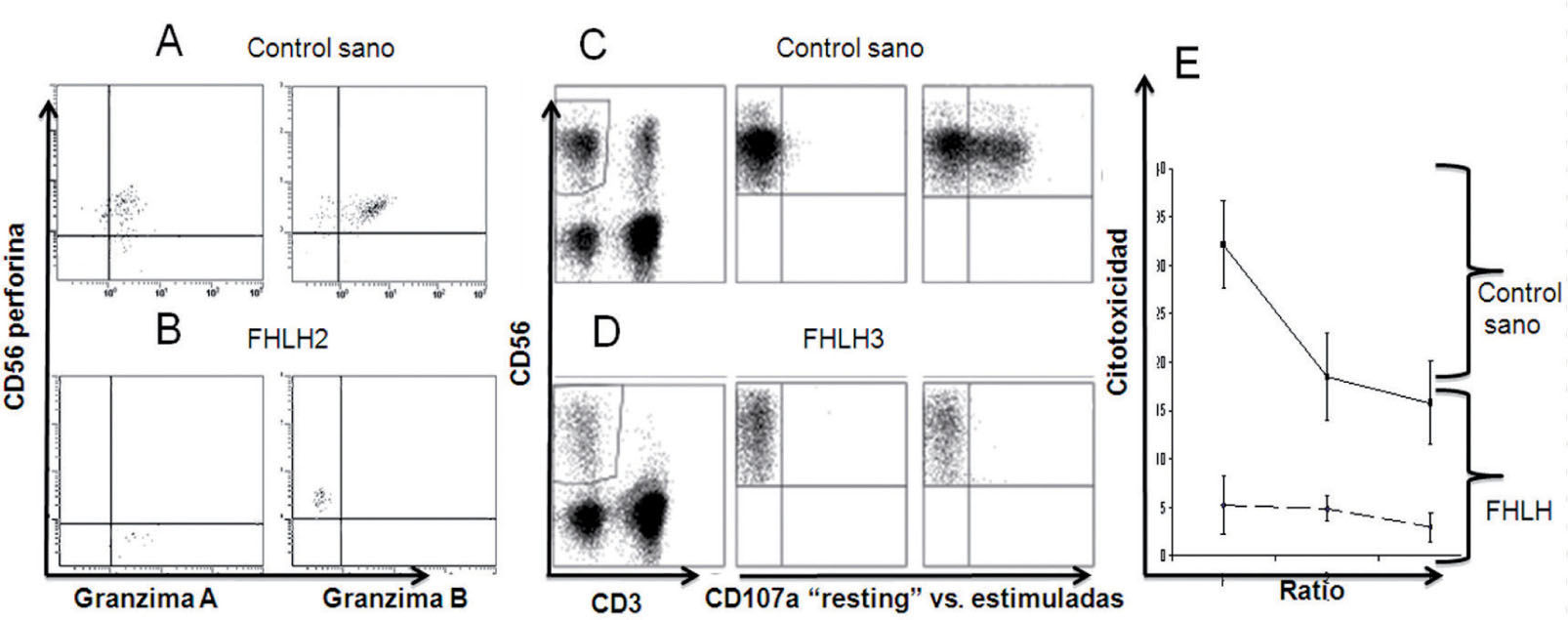
 y un paciente con FHLH2 (B). Ensayo de degranulación, expresión de CD107a, en un control sano (C) y en un paciente con FHLH3 (D). Ensayo de citotoxicidad in vitro de las células NK frente a la línea celular K562, donde se observa la disminución de la misma en el paciente con FHLH en comparación con el control sano (E). FHLH: linfohistiocitosis hemofagocítica familiar.")
Expresión de perforina y granzima por citometría de flujo en un control sano (A) y un paciente con FHLH2 (B). Ensayo de degranulación, expresión de CD107a, en un control sano (C) y en un paciente con FHLH3 (D). Ensayo de citotoxicidad in vitro de las células NK frente a la línea celular K562, donde se observa la disminución de la misma en el paciente con FHLH en comparación con el control sano (E).
FHLH: linfohistiocitosis hemofagocítica familiar.
Las pruebas genéticas constituyen la prueba de oro para el diagnóstico de HLH. Sin embargo, son costosas, laboriosas, accesibles para pocos grupos y, en ocasiones, de difícil interpretación. Las pruebas genéticas deben incluir el análisis mutacional de PRF1, UNC13D, STX11 y Munc18-212,13. En los pacientes varones con antecedentes o historia familiar de trastornos linfoproliferativos, linfomas o infección complicada por virus de Epstein-Barr (VEB) debe incluirse además el análisis de los genes SH2D1A y BIRC4. La confirmación del diagnóstico genético implica el estudio de extensión a los hermanos para descartar la enfermedad o determinar su idoneidad como potencial donante para el TPH, además de ayudar a establecer un adecuado consejo genético. La mayoría de las mutaciones en homocigosis originan un cuadro clínico grave; sin embargo, algunos defectos genéticos monoalélicos también pueden producir fenotipos clínicos muy graves, como por ejemplo las mutaciones en heterocigosis para PRF1. Además, FHLH puede ser poligénica y afectar en heterocigosis a mutaciones en PRF1 y UNC13D, por lo que es necesario identificar los diferentes genes alterados.
En ocasiones, el diagnóstico genético es complejo y no es suficiente con la secuenciación del exoma. Existen mutaciones en intrones, en zonas de splicing o polimorfismos de un solo nucleótido que dificultan el diagnóstico, sobre todo en los casos de presentación clínica atípica, edad avanzada y en algunas etnias.
Aproximadamente en el 50% de los pacientes pediátricos con HLH no se encuentra un defecto genético conocido14. En estos casos, sospecharemos una forma primaria si existe historia familiar, si el cuadro clínico es grave, recurrente o persiste la actividad citotóxica NK disminuida. En estos casos, el tratamiento no debe retrasarse aun en ausencia del diagnóstico genético.
Biomarcadores: nuevas herramientas diagnósticasLos marcadores biológicos permiten la evaluación en curso de la actividad de los CTL, NK y macrófagos. Estas pruebas pueden identificar la gravedad de la enfermedad, el pronóstico y la respuesta al tratamiento, e incluso puede ser útil en la predicción de la recaída HLH. Los niveles de sCD25 se correlacionan con la activación de los linfocitos T y tienen una gran importancia pronóstica. Este receptor es una proteína de transmembrana que se sobreexpresa cuando los linfocitos T están activados, por lo que el dominio extramembrana se desprende de la superficie celular conservando su capacidad para unirse a IL-2. Las altas concentraciones séricas de sCD25 se han descrito en la HLH y en el síndrome de activación macrofágica (MAS). La proteína CD163 es una proteína expresada en la superficie de los monocitos y macrófagos, que se sobreexpresa cuando los monocitos y macrófagos están activados. Al igual que sCD25, el dominio extracelular de CD163 puede liberarse y hacerse soluble (sCD163). La combinación de sCD25 y sCD163 puede ser útil en el diagnóstico, en la respuesta al tratamiento y en el seguimiento de la actividad de la enfermedad15.
Recientemente, Bryceson et al. han descrito una población de linfocitos CD57+, representando una subpoblación de CTL con mayor capacidad de degranulación, por lo que la selección de esta población para su estudio molecular podría ayudar al diagnóstico de FHL con déficit en la degranulación (FHL-3, 4 y 5)16.
En paralelo a las pruebas inmunológicas, debe realizarse un cuidadoso estudio microbiológico para identificar desencadenantes infecciosos, en particular VEB, citomegalovirus y Leishmania. El tratamiento de estas causas infecciosas es un elemento indispensable para el adecuado tratamiento de la HLH.
Trastornos metabólicos característicos de linfohistiocitosis hemofagocíticaAlteraciones en los niveles séricos de ferritina, fibrinógeno y/o triglicéridos pueden encontrarse en pacientes con HLH. La ferritina es una proteína implicada en la homeostasis del hierro y generalmente se encuentra elevada en estados de inflamación como reactante de fase aguda. Como tal, es un componente importante en la toma un diagnóstico clínico de HLH. Valores superiores a 500μg/l son indicativos y valores superiores a 10.000 μg/l prácticamente exclusivos de HLH. La disminución de este parámetro tras el inicio del tratamiento se relaciona con una buena respuesta. La hipofibrinogenemia grave es típica de HLH, y en asociación con trombopenia y coagulopatia es muy indicativa. La etiología de la hipertrigliceridemia está poco clara, aunque suele asociarse a disfunción hepática. Otros reactantes de fase aguda, como la velocidad de sedimentación de los eritrocitos o la proteína C reactiva, se pueden utilizar para controlar la actividad de la enfermedad17,18.
Diagnóstico diferencialLa HLH es un síndrome que agrupa diferentes enfermedades con datos clínicos y de laboratorio similares. Como hemos mencionado previamente, la HLH se clasifica en 2 categorías de acuerdo con las etiologías subyacentes: primarios y secundarios. Las formas primarias se heredan de forma autosómica recesiva o ligada al cromosoma X, o bien forman parte de alguna inmunodeficiencia. Las formas secundarias ocurren en pacientes sin defectos genéticos conocidos y asocia factores desencadenantes. Estos factores son las infecciones, fundamentalmente virales y las enfermedades autoinmunes, fundamentalmente en la artritis reumatoide juvenil sistémica. Otros desencadenantes pueden ser las neoplasias, fundamentalmente las leucemias linfoides, y los tratamientos inmunosupresores y quimioterápicos. Dada la heterogeneidad del diagnóstico de HLH, es necesario buscar la causa subyacente.
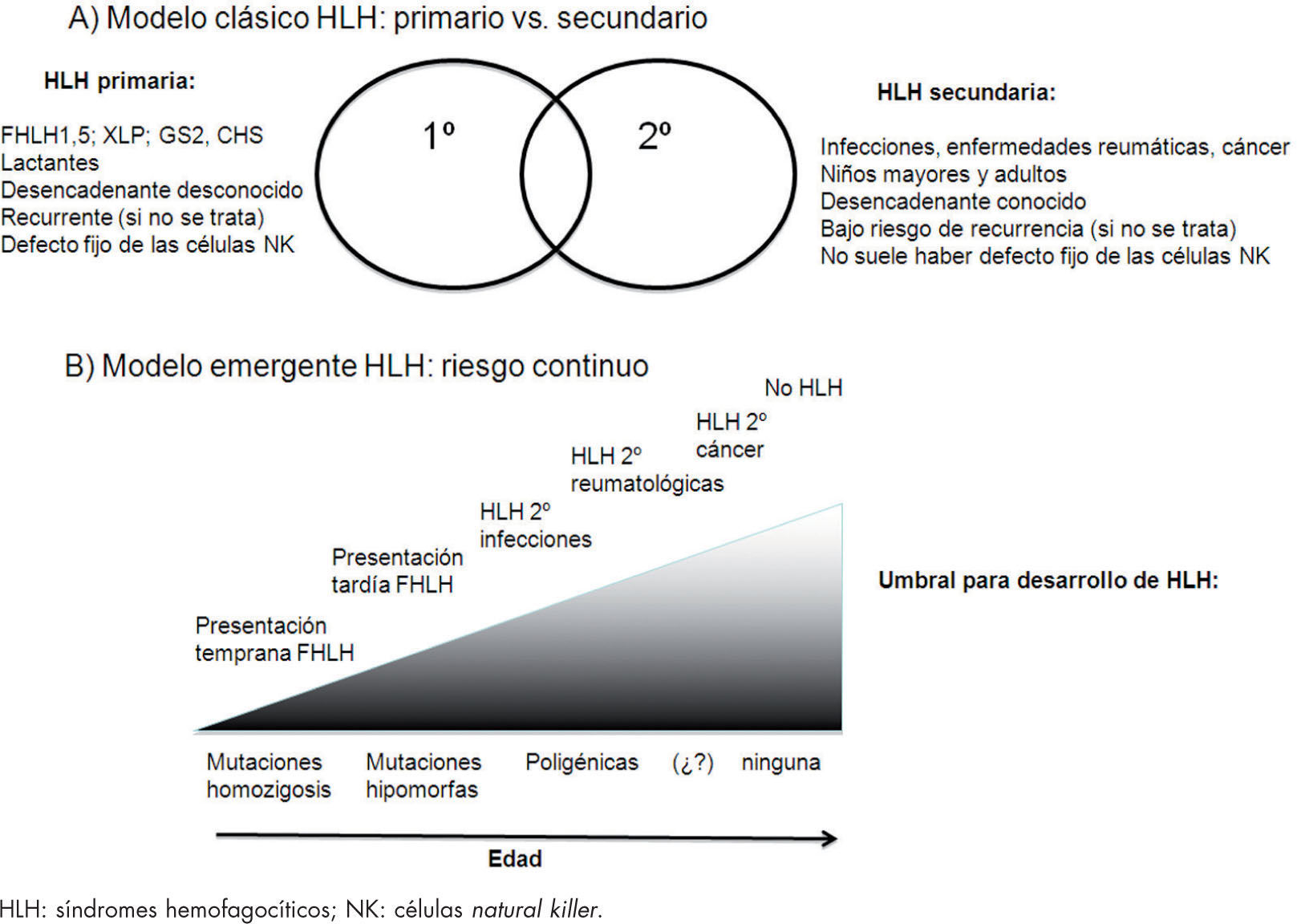
Pacientes con sepsis, síndrome de respuesta inflamatoria sistémica y síndrome de disfunción multiorgánica, a menudo cumplen los criterios diagnósticos de HLH. La fiebre, las citopenias, la hipertrigliceridemia, la hipofibrinogenemia y los altos niveles de ferritina son comunes a estas 3 entidades. Incluso la hemofagocitosis en médula ósea o en cualquier otro órgano ha sido descrita. La elevación de sCD25 y la disminución de la actividad citotóxica de las células NK, hallazgos indicativos de HLH, se han descrito también en la sepsis, por lo que el diagnóstico diferencial de estas entidades resulta clave para evitar tratamientos inadecuados que puedan empeorar el cuadro clínico19. Recientemente, algunos autores señalan que estas afecciones constituyen diferentes formas clínicas de una misma forma continua de disfunción del sistema inmunitario que origina un estado de hiperinflamación sistémica. En presencia de mutaciones genéticas que originen una extensa disfunción de las células efectoras los pacientes desarrollarían FHLH; sin embargo, las alteraciones genéticas menores, como polimorfismos, que originen defectos parciales originarían formas secundarias. Estos datos son importantes, porque podría ser beneficioso el tratamiento inmunosupresor en pacientes con sepsis grave o shock séptico en quienes la respuesta inmunitaria es la responsable del daño tisular permanente (fig. 3)20–22.
 HLH primarios, enfermedad genética de herencia autosómica recesiva o ligada al cromosoma X, con agrupación familiar y aparición en niños muy pequeños frente a HLH secundarios, trastornos esporádicos en niños mayores sin alteraciones genéticas conocidas. B) HLH como síndrome único con diferentes expresiones en función de las diferentes alteraciones genéticas (mutaciones completas, hipomorfas, poligénicas, polimorfismos) que asocian un riesgo continuo de desarrollar HLH en respuesta a diferentes estímulos inmunitarios leves o extremos. HLH: linfohistiocitosis hemofagocítica.")
Modelos conceptuales de HLH. A) HLH primarios, enfermedad genética de herencia autosómica recesiva o ligada al cromosoma X, con agrupación familiar y aparición en niños muy pequeños frente a HLH secundarios, trastornos esporádicos en niños mayores sin alteraciones genéticas conocidas. B) HLH como síndrome único con diferentes expresiones en función de las diferentes alteraciones genéticas (mutaciones completas, hipomorfas, poligénicas, polimorfismos) que asocian un riesgo continuo de desarrollar HLH en respuesta a diferentes estímulos inmunitarios leves o extremos. HLH: linfohistiocitosis hemofagocítica.
Las formas primarias de HLH presentan una evolución rápida y fatal en pocas semanas en ausencia del tratamiento adecuado, por lo que este debe instaurarse precozmente, si existe una alta sospecha diagnóstica, incluso con ausencia del resultado de pruebas confirmatorias. La presencia de citopenias, disfunción orgánica o infecciones no resueltas no son limitaciones para iniciar el tratamiento23,24. El principal objetivo terapéutico es suprimir la inflamación e inducir apoptosis de CTL, NK, macrófagos y APC. Para ello, utilizaremos fundamentalmente esteroides, dexametasona preferiblemente por su capacidad de penetrar en el sistema nervioso central (SNC), y VP16, un quimioterápico potente que induce apoptosis celular. La dosis de VP16 debe reducirse si existen datos de insuficiencia renal junto con hiperbilirrubinemia directa. No debe reducirse la dosis en los casos de hiperbilirrubinemia aislada o neutropenia. En los pacientes con afectación del SNC se debe instaurar tratamiento intratecal con esteroides y metotrexato. La ciclosporina ha sido relegada a etapas posteriores del tratamiento por la dudosa efectividad del mismo y su elevada toxicidad, fundamentalmente neurológica.
Este esquema terapéutico está recogido en el protocolo HLH-2004, que consta de una fase de inducción a la remisión de 8 semanas que incluye etopósido, dexametasona, con/sin hidrocortisona/metotrexato intratecal semanal, seguida de una fase de mantenimiento de hasta 40 semanas con pulsos de dexametasona, VP16 y ciclosporina (en ausencia de nefropatía, hipertensión arterial, hepatopatía y alteraciones neurológicas)2. Sin embargo, el tratamiento de mantenimiento puede prolongarse el tiempo necesario hasta que el TPH esté dispuesto. La terapia intratecal debe reservarse para los casos donde no mejoren los síntomas neurológicos, incluidas anomalías en el líquido cefalorraquídeo tras 2 semanas del inicio del tratamiento.
Con este esquema, alrededor del 50–75% de los pacientes entran en remisión.
Algunos autores han descrito resultados similares utilizando globulina antitimocito (ATG) y ciclosporina, con menos complicaciones que las reportadas con VP16. Sin embargo, la recomendación es reservar la ATG para los casos en los que no pueda utilizarse el VP16 (hiperbilirrubinemia con insuficiencia renal).
En las formas refractarias, el tratamiento no está claro. Se sugiere utilizar fármacos de segunda línea como ATG, alentuzumab (anti-CD52), daclizumab, vincristina y antifactor de necrosis tumoral alfa (TNF-α) (etanercept e infliximab)25–28.
Los pacientes con HLH secundaria que no mejoran con tratamiento etiológico, o con una presentación clínica grave, puede beneficiarse del protocolo HLH-2004, interrumpiendo el mismo tras 8 semanas de tratamiento. En estos casos, solo se indicará TPH si la enfermedad es recurrente o persistente, incluso en ausencia de diagnóstico genético2.
En los pacientes diagnosticados de MAS, el tratamiento con dosis de esteroides altas y/o con ciclosporina A mejora los síntomas en pocos días en más de la mitad de los pacientes. El VP16 puede añadirse en las formas refractarias o en presencia de mucha activación. El control de la enfermedad reumatológica es muy importante, por lo que el tratamiento de base de la enfermedad con antagonistas de los receptores IL-1/IL-6 y anti-TNF-α debe mantenerse. Sin embargo, estos tratamientos pueden desencadenar HLH y agravar el cuadro.
La HLH secundaria por VEB puede tratarse con rituximab (anti-CD20) e inmunoglobulinas junto con la terapia convencional, alcanzando hasta un 75% de remisiones. El VP16 debe reservarse para los pacientes refractarios y recurrentes. Un 15% de los pacientes con HLH secundario a VEB requieren el TPH. Recientemente, se ha indicado que el alemtuzumab puede ser útil como fármaco de rescate en estos pacientes28,29.
El tratamiento de soporte es fundamental en estos pacientes y deben recibir profilaxis para infecciones oportunistas con cotrimoxazol, fluconazol e inmunoglobulinas. Además, deben recibir los cuidados habituales de los pacientes neutropénicos.
El trasplante de progenitores hematopoyéticosEl TPH se recomienda en las formas primarias, en las formas secundarias recurrentes en ausencia de diagnóstico genético, o que progresan pese al tratamiento adecuado, y en los pacientes con persistencia de disfunción citotóxica de las células NK. Algunos autores indican que los casos severos de HLH asociados a VEB deberían trasplantarse30. Actualmente, el TPH es el único tratamiento curativo para los pacientes con alteraciones genéticas, y debe realizarse pronto, tras el control de la enfermedad, por lo que es necesario iniciar la búsqueda de un donante tan pronto como el diagnóstico de HLH primaria se establezca. Tradicionalmente, se han utilizado regímenes de acondicionamiento mieloablativo, que provocaban síndrome de obstrucción sinusoidal y neumonitis no infecciosa, con una elevada mortalidad. El desarrollo de acondicionamientos de intensidad reducida basados en inmunosupresores como fludarabina o alentuzumab han permitido disminuir estas complicaciones31–34. Las principales limitaciones de este tipo de TPH son el quimerismo hematopoyético mixto y el riesgo de fallo secundario del injerto. Sin embargo, hasta un 10–15% de quimerismo donante puede ser suficiente para mantener la remisión de la enfermedad y evitar la recurrencia16. En estos casos, la monitorización del quimerismo y de las pruebas inmunológicas resulta clave para instaurar diferentes tratamientos, como la retirada de la inmunosupresión, infusiones de progenitores hematopoyéticos o la infusión de linfocitos.
La selección del donante familiar puede resultar problemática, especialmente en los casos donde desconocemos la genética subyacente. La posibilidad de utilizar un hermano enfermo como donante se incrementa por el hecho de que la edad de presentación puede ser variable entre hermanos. En estos casos, la actividad citotóxica NK puede ser un parámetro de selección, eligiendo los familiares idénticos con actividad NK normal, o sugerir un donante no familiar, como mejor opción35.
El papel fundamental del interferón gamma (IFN-γ), tal y como describimos en la fisiopatología anteriormente, indica una atractiva diana terapéutica a desarrollar a corto plazo en ensayos clínicos con pacientes con HLH.
PronósticoA pesar de haber mejorado la supervivienda de estos pacientes desde el inicio del protocolo HLH-94, todavía el 40% de los pacientes fallecen durante el tratamiento de inducción por progresión de la enfermedad o por infecciones intercurrentes. Se consideran datos de mal pronóstico al diagnóstico la hiperbilirrubinemia, la trombopenia, la hiperferritinemia y la pleocitosis en el líquido cefalorraquídeo. Tras el tratamiento, se consideran datos de mal pronóstico la persistencia de la anemia, la trombopenia, la hipofibrogenemia, la disfunción hepática y la elevación del sCD25, así como la persistencia de la fiebre. En las HLH inducidas por VEB, una elevada carga viral al diagnóstico y la persistencia de la misma tras el tratamiento se consideran de mal pronóstico. Los pacientes que desarrollan insuficiencia respiratoria con infiltrados pulmonares presentan un pronóstico muy desfavorable.
Antes del TPH, es necesario que la enfermedad se encuentre inactiva y sin signos de afectación del SNC. Aproximadamente, el 70% de los pacientes con formas primarias son trasplantados y alcanzan supervivencias a largo plazo del 50–60%. El TPH en las formas secundarias alcanza supervivencias a los 5 años del 86% cuando el desencadenante es infeccioso, e inferiores al 15% cuando la causa es tumoral. Las complicaciones a largo plazo de HLH tras el TPH son fundamentalmente derivadas del propio procedimiento; sin embargo, no son excepcionales las reactivaciones en SNC, por lo que la monitorización mediante neuroimagen y análisis del líquido cefalorraquídeo resultan fundamental en el período postrasplante.
El pronóstico de las formas secundarias es incierto y debemos establecer un cuidadoso diagnóstico diferencial con otras formas clínicas, como el shock séptico y el síndrome de disfunción múltiple orgánica. Los nuevos conocimientos muestran que nos encontramos ante diferentes formas clínicas de un mismo espectro patológico.
La complejidad y la gravedad de la HLH aconsejan que su manejo se realice en centros con experiencia y donde puedan articular las diferentes herramientas diagnósticas (inmunológicas y genéticas) y terapéuticas (TPH). El conocimiento creciente de las bases genéticas de la HLH resulta clave para el desarrollo futuro de nuevas terapias génicas, actualmente en desarrollo en modelos animales con resultados preliminares satisfactorios.
La mejora del diagnóstico genético permitirá, a medio plazo, optimizar el consejo genético y el diagnóstico precoz prenatal y preimplantacional, así como adecuar la idoneidad de los donantes. Probablemente, la terapia génica sustituirá al TPH para curar las formas genéticas conocidas.