




Los tumores del cuerpo carotídeo son tumores raros, muy vascularizados, originados en los quimiorreceptores de la bifurcación carotídea. Se ha descrito su malignización y asociación con otros tumores endocrinos. Pueden ser familiares en el 10-50% de los casos.
ObjetivoAnalizar nuestros resultados en el manejo de estos tumores y presentar un protocolo de actuación y seguimiento.
Material y métodosEntre 1986 y 2008 se trataron 25 casos en 23 pacientes (2 bilaterales), con una edad media de 51 años (r 31-78), el 82,6% mujeres, un caso familiar. Como pruebas diagnósticas se realizaron ecodoppler y arteriografía en todos los pacientes, y tomografía computarizada o resonancia magnética en 12. El seguimiento postoperatorio se llevo a cabo con ecodoppler.
ResultadosEn todos los casos se realizó la resección completa del tumor, los 14 últimos con embolización preoperatoria. Fue necesaria la reconstrucción vascular en 6 casos. Seis pacientes tuvieron lesión neurológica, uno rotura arterial y uno neumonía postoperatoria. Según los criterios de clasificación de Shamblin, 13 casos fueron tipo I, 5 tipo II y 7 tipo III. Las complicaciones y reconstrucciones vasculares estuvieron relacionadas con tumores tipo II y III. Durante el seguimiento se detectaron 4 recidivas, 2 tumores contralaterales (bilaterales), un feocromocitoma, una trombosis del injerto y 6 éxitus (uno por malignización y metástasis).
ConclusionesLa cirugía en fase precoz disminuye la morbimortalidad. El seguimiento con ecodoppler permite detectar recidivas, bilateralidad y complicaciones de la reconstrucción vascular. La posible presentación familiar, recidivas, asociación a otros tumores o metástasis, hacen necesario el seguimiento sistematizado del paciente.
Carotid body tumours are rare, highly vascular, arising from the chemoreceptors of the carotid bifurcation. Their relationship with malignancy and other endocrine tumours have already been described. There can be between 10% and 50% of cases in a family.
ObjectiveTo analyse the results in the management of these tumours, and present a protocol for action and monitoring.
Material and methodsBetween 1986 and 2008 a total 25 cases were treated in 23 patients (2 bilateral) with a mean age of 51 years (range 31-78), with 82.6% women, 1 family case. Doppler ultrasound and angiography were performed as diagnostic tests in all patients, with computed tomography or magnetic resonance imaging in 12 cases. They were monitored annually using Doppler ultrasound.
ResultsAccording to the Shamblin classification criteria, 13 cases were type I, 5 were type II and 7 type III. A complete resection of the tumour was performed in all cases, the last 14 with preoperative embolisation. Vascular reconstruction was necessary in 6 cases. Six patients had neurological injury, an arterial rupture and a postoperative pneumonia. Complications and vascular reconstruction were associated with type II and III tumours. During follow-up four recurrences were detected, two contralateral tumours, a phaeochromocytoma, a graft thrombosis and six deaths, (1 from malignancy and metastasis).
ConclusionsSurgery in the early stage reduces the morbidity and mortality. Follow-up should be with Doppler ultrasound to detect recurrence, bilaterality and complications of vascular reconstruction. The possible familial presentation, recurrence, association with other tumours or metastases, requires systematic monitoring of the patient.
Los quemodectomas, o paragangliomas, son tumores neuroendocrinos que derivan de la cresta neural o neuroectodermo y se clasifican en suprarrenales (feocromocitomas) y extrasuprarrenales (carotídeo, yugular, vagal, aórtico, autónomo visceral etc.). Los quemodectomas o tumores del cuerpo carotídeo, dentro de su rareza, son los paragangliomas extrasuprarrenales más frecuentes1. Se originan en los quimiorreceptores de la bifurcación carotídea, son tumores muy vascularizados y de crecimiento lento que pueden englobar y comprimir estructuras vasculares y nerviosas. La forma más frecuente de presentación es como masa laterocervical asintomática y pueden ser bilaterales. La mayoría son esporádicos, la forma familiar está descrita en el 10-50% de los casos2,3. Generalmente son benignos, aunque pueden malignizarse y dar metástasis a distancia (pulmonar, hepática, ósea). Ocasionalmente se asocian a otros tumores neuroendocrinos. El objetivo de este estudio es mostrar nuestra experiencia en el manejo de estos tumores y presentar un protocolo de actuación y seguimiento en pacientes y familiares.
Material y métodosRealizamos una revisión retrospectiva de 25 tumores del glomus carotídeo tratados en nuestro servicio en 23 pacientes durante un periodo de 22 años (1986- 2008); 2 de ellos (8%) fueron bilaterales.
La edad media de presentación fue de 51 años (r 31-78), 4 varones (7,4%) y 19 mujeres (82,6%). La forma de presentación fue como masa laterocervical palpable en 21 casos (84%). En 10 pacientes (43,5%) hubo antecedentes personales de hipertensión arterial, en 3 (13%) isquemia cerebrovascular y en 2 (8,7%) tiroidectomía. Un caso fue familiar (4,35%). Para el diagnóstico se realizaron ecodoppler y arteriografía en todos los pacientes. En 7 se completó el estudio con tomografía computarizada (TC) cervical para determinar la extensión del tumor debido a su gran volumen y en 5 pacientes ya se había realizado el diagnóstico con resonancia magnética (RM) antes de ser remitidos a nuestras consultas. El seguimiento postoperatorio lo realizamos con ecodoppler carotídeo bilateral anual, y cada 2 años al superar los 10 años de seguimiento postoperatorio sin detección de incidencias. En caso de sospecha de recidiva realizamos angioTC o RM
ResultadosSe realizó la resección completa del tumor en los 25 casos, con embolización preoperatoria en los 14 últimos casos de nuestra serie (56%). La embolización se practicó con micropartículas de polivinilalcohol 24-48h antes de la cirugía, y como complicaciones de la misma un paciente (7%) tuvo un AIT con monoparesia braquial y otro (7%) un hematoma pulsátil femoral que se resolvió con inyección de trombina.
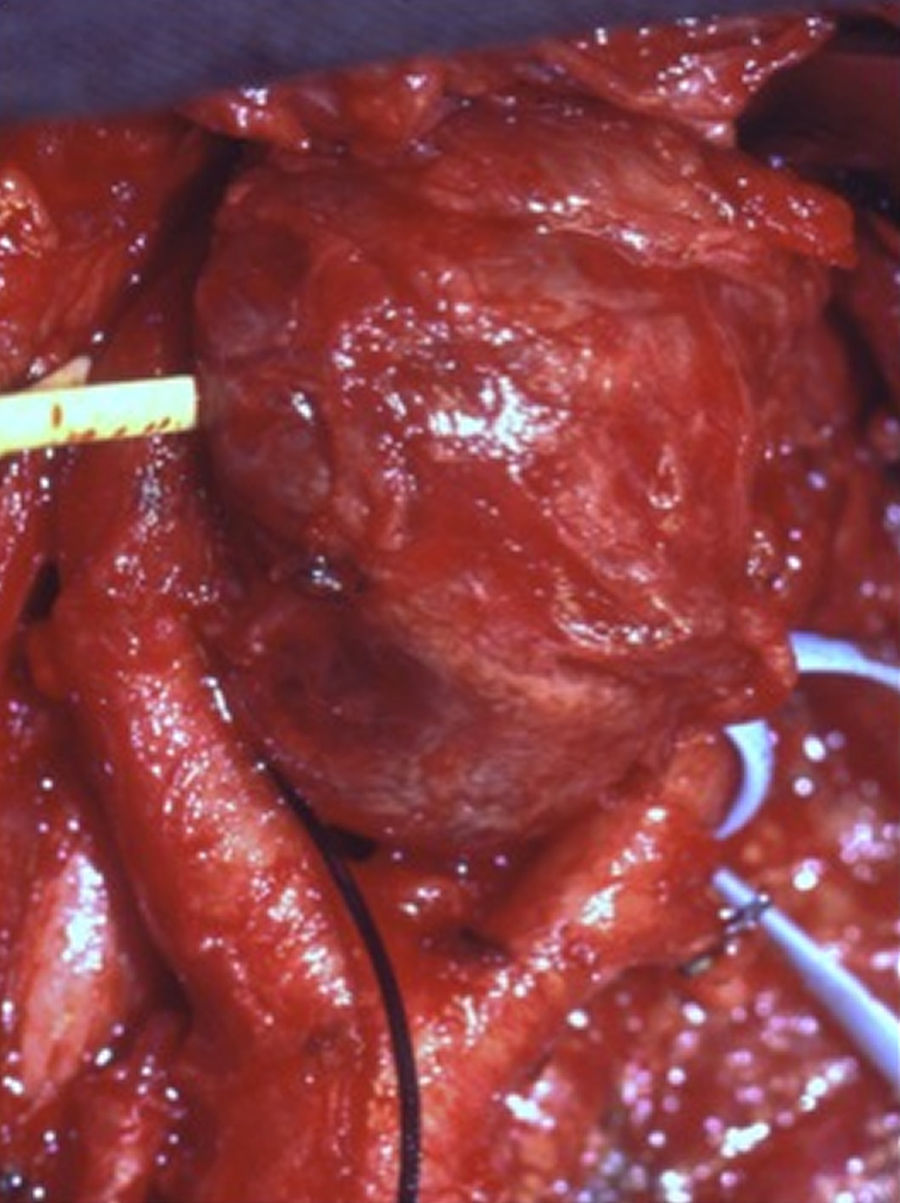
Los tumores se clasificaron según los criterios establecidos por Shamblin: 13 casos tipo I (52%), 5 casos tipo II (20%) y 7 casos tipo III (28%) (fig. 1). En todos se realizó confirmación anatomopatológica histológica e inmunohistoquímica.
. Impronta de la bifurcación carotidea en polo inferior de quemodectoma.")
Todos los casos se realizaron con anestesia general y mediante cervicotomía longitudinal por delante del esternocleidomastoideo. Hubo dificultad en el control de la tensión arterial durante la cirugía en 2 casos (8%) y 3 casos (12%) requirieron transfusión sanguínea (Shamblin tipo III). Un paciente (4%) precisó traqueotomía debido a las dificultades para la intubación por compresión de la vía aérea por el tumor (Shamblin tipo III) y fue necesario ampliar la incisión (en sentido cefálico hasta la mastoides) en sentido ascendente hasta el borde anterior del orificio auditivo externo.
Seis casos (24%) precisaron reconstrucción vascular (5 tipo III y uno tipo II); en 5 de ellos (83%) se reconstruyó la carótida interna: 2 casos mediante sutura término-terminal, 2 resecciones en bloque del tumor y de la carótida e interposición de injerto de safena interna y en uno se realizó endarterectomía carotídea y cierre con parche biológico de pericardio bovino Vascuguard® (por rotura y sangrado tras pico hipertensivo una vez resecada la tumoración). En un caso (16%) se reparó la carótida externa con sutura término-terminal.
No hubo mortalidad precoz. Como complicaciones neurológicas, 5 pacientes (20%) tuvieron disfonía transitoria por lesión del X par craneal. En la tabla 1 se ve la relación de estas complicaciones con la clasificación de Shamblin. Un caso (4%) presentó neumonía postoperatoria que se resolvió con tratamiento antibiótico. Recidivaron 4 casos (16%), 3 Shamblin tipo I y uno tipo III, de los cuales se volvieron a resecar 2, uno se embolizó y otro está pendiente de tratamiento. Las recidivas se detectaron a los 13, 48, 144 y 224 meses de la cirugía (al año, 4, 12 y 19 años de seguimiento). El caso detectado dentro del primer año de la cirugía podría explicarse por la permanencia de células tumorales (microscópicas) y se llevó a cabo la embolización a los 15 meses como único tratamiento. En uno de los casos reintervenidos se produjo lesión del nervio hipogloso y síndrome de Claude Bernard Horner. Este último caso se trata de una mujer de 33 años que presentó paragangliomas múltiples. Fue intervenida de glomus carotídeo bilateral, de feocromocitoma epicárdico y de un feocromocitoma vesical que había comenzado con crisis hipertensivas y síncopes miccionales. Recientemente ha sido intervenida también de un meningioma cerebral y presenta recidiva del tumor de glomus carotídeo izquierdo. El estudio genético determinó que es portadora de una mutación del gen SDHB (1p36). En el seguimiento, aparte de las 4 recidivas mencionadas anteriormente, se detectó un caso de tumor contralateral (4%), uno de trombosis asintomática del injerto (4%), y un caso asociado a otros tumores (4%) (el caso ya mencionado de paragangliomas múltiples). Fallecieron por causas no relacionadas con el tumor 5 pacientes (21,7%) y uno (4,3%) por malignización del tumor. En el caso de malignización, se detectó la recidiva del tumor y la existencia de metástasis óseas y pulmonares 4 años después de la cirugía en una TC toracoabdominal. La recidiva fue reintervenida y, para tratar las metástasis, oncología aplicó un ciclo de quimioterapia con cisplatino-vulmon-silmetan. pero la paciente falleció por enfermedad pulmonar en el contexto de su enfermedad de base diseminada.
Morbilidad relacionada con la clasificación de Shamblin
| Casos quirúgicos (27) | Shamblin | Reconstrucción vascular | Lesión neurológica |
| Primarios (25) | I (13) | 0 | 1 (3,7%) |
| II (5) | 1 (3,7%) | 2 (7,4%) | |
| III (7) | 5 (18,5%) | 3 (11,1%) | |
| Recidivados (2)a | I (1) | 0 | 0 |
| III (1) | 0 | 1 (3,7%) |
Los paragangliomas de cabeza y cuello son raros, con una incidencia de 1:30.000-1:100.000 habitantes, siendo el más frecuente el tumor del glomus o cuerpo carótideo (78%)4. Histológicamente es un tumor incompletamente encapsulado, muy vascularizado y formado por 2 tipos de células.
Solo el 5% tiene actividad endocrina y puede producir hipertensión; es más frecuente la secreción de catecolaminas en los paragangliomas retroperitoneales2. La mayor parte son tumores benignos, de crecimiento lento, pero algunos pueden malignizar. El potencial maligno no se establece por la anatomía patológica, solo puede establecerse por la recurrencia local, la extensión a tejidos contiguos y la metástasis por vía linfática y hematógena a ganglios regionales, pulmón, huesos y corazón. La incidencia de metástasis local o a distancia es de menos del 10%5,6. En nuestra serie, un caso (4,35%) presentó metástasis pulmonares y óseas.
Otro aspecto característico es que pueden ser multicéntricos, dependiendo de su localización en tejido con actividad simpática (incluidos quimiorreceptores, médula suprarrenal y ganglios retroperitoneales), por lo que es difícil determinar con precisión su comportamiento biológico o la asociación a neoplasias endocrinas múltiples7. Los mejores métodos para detectar tumores múltiples son la TC o la RM7, y en casos con actividad funcional la gammagrafía marcada con metaiodobencilguanidina (MIBG 123I)8. Lo que no está claro es con qué periodicidad habría que realizar estos estudios.
Es importante el diagnóstico y resección quirúrgica precoz del tumor, ya que cuando son pequeños están poco adheridos y son fácilmente extirpables, con una menor incidencia de complicaciones9. En nuestra serie, los 6 casos (24%) de reconstrucciones vasculares como los 6 casos (24%) de complicaciones neurológicas estuvieron relacionadas con tumores tipos II y III de la clasificación de Shamblin (tabla 1), similar a lo evidenciado por otros grupos10,11.
Existe controversia en cuanto a la embolización preoperatoria de estos tumores. Algunos estudios señalan que la embolización facilita la cirugía, ya que disminuye la pérdida de sangre, el tiempo operatorio y la morbilidad por lesión neurológica12–14; otros estudios no encuentran diferencias entre la pérdida de sangre, el tiempo quirúrgico y la morbilidad perioperatoria12,13,15. El riesgo de accidente cerebrovascular se ha descrito superior al 10%13,14,16,17. En nuestra experiencia, desde el año 1997 realizamos de forma sistemática embolización preoperatoria en todos los tumores, ya que nos resulta más fácil la resección del tumor por menor sangrado, y tan solo hemos tenido 2 complicaciones leves con la embolización (un AIT y un hematoma femoral que se resolvió con inyección de trombina).
Las formas familiares están relacionadas con una mutación en el gen SDHB, SDHC, o SDHD, (succinato deshidrogenasa subunidades B, C y D), siendo la mayoría por mutaciones en el gen SDHD. En el caso de SDHD, solo la transmisión por vía paterna lleva a la génesis de tumores en la descendencia, pero también se ha observado que el desarrollo de los tumores en portadores se incrementa en altitudes elevadas. La SDHD tiene imprinting materno: si la mutación se recibe de la madre no se expresará, pero puede transmitirse, y en el caso de que lo haga un varón, sí puede tener descendencia afectada18. Trabajos recientes han localizado el gen en la región 11q 23q lo cual podrá permitir la pesquisa genética en individuos con riesgos19. Cuando los quemodectomas son familiares aparecen múltiples o bilaterales con mucha mayor frecuencia que cuando son esporádicos (31,8% vs 4,4%)20–22. En nuestra serie de 23 pacientes tan solo se realizó estudio genético en la paciente con paragangliomas múltiples, que resultó tener una mutación del gen SDHB (1p36). Fakhry et al.23 encuentran mutación genética en 8 de 23 pacientes estudiados (35%), la mayoría en el gen SDHD y el 87% tuvieron paragangliomas en otras localizaciones, por eso recomiendan estudio genético en todos los pacientes con paragangliomas cervicales. Actualmente realizamos estudio genético sistemáticamente a todos los pacientes nuevos diagnosticados de tumor del glomus carotídeo.
El seguimiento de nuestros pacientes se hace mediante exploración física y ecodoppler carotídeo anual. El seguimiento con ecodoppler permite detectar recidivas, bilateralidad y complicaciones en casos de reconstrucción vascular. No obstante, debido a las peculiares características de estos tumores, nos vemos obligados a plantearnos realizar un despistaje familiar, buscar tumores neuroendocrinos asociados, metástasis en algunos casos y posible bilateralidad, y a valorar la posible funcionalidad del tumor. Todo esto hace necesario un seguimiento sistematizado, y al tratarse de una patología multidisciplinar proponemos un protocolo en el que intervendrían los Servicios de Radiología Vascular, Endocrinología y la Unidad de Genética.
En el protocolo que proponemos, el Servicio de Angiología y Cirugía Vascular, enviará a la Unidad de Genética a todos los pacientes diagnosticados de tumor del glomus, para estudio y asesoramiento genético. En caso de presentar alguna mutación genética, se recomendaría el estudio de los familiares de primer grado. No debemos olvidar los aspectos éticos que implica el padecer una alteración o enfermedad genética. Es imperativo respetar la intimidad, confidencialidad y la decisión autónoma del paciente de someterse o no a estudios genéticos que le podrían afectar de por vida psicológica y laboralmente (estigmatización del paciente y sus familiares, rechazo de empresas o compañías de seguros); esto conlleva nuevas e importantes connotaciones éticas que se plantean a los profesionales y a la sociedad. Por otra parte, en colaboración con el Servicio de Endocrinología se debe determinar la posible funcionalidad del tumor mediante la determinación de catecolaminas en orina y descartar tumores asociados mediante TC o RM de abdomen y pelvis y gammagrafia con MIBG 123I (tabla 2).
Protocolo
| Protocolo de diagnóstico y tratamiento |
| Anamnesis personal y familiar y exploración |
| Ecodoppler+angioTC cervical |
| Arteriografía |
| Embolización preoperatoria |
| Tratamiento quirúrgico en 24-48 h |
| Estudio anatomopatológico del tumor |
| Estudio molecular de los genes SDHB, SDHC, SDHD en muestra de sangre periférica |
| Determinación de catecolaminas en orina |
| Protocolo de seguimiento de pacientes y familiares |
| Pacientes sin mutación genética (esporádicos) |
| Ecodoppler anual |
| Consultas de endocrino |
| Catecolaminas en orina anual |
| TC/RM abdomino-pélvica: cada 2-3 años |
| Gammagrafía con MIBG 123I cada 2-3 años |
| Pacientes con mutación genética |
| Ecodoppler anual |
| Consultas de endocrino |
| Catecolaminas en orina anual |
| TC/RM abdomino-pélvica anual |
| Gammagrafía con MIBG 123I anual |
| Estudio genético familiares de primer grado |
| Familiares portadores de la mutación |
| Ananmnesis y exploración, y ecodoppler anual |
| TC/RM cervical: si se sospecha tumor por ecodoppler |
| Consultas de endocrino |
| Catecolaminas en orina anual |
| TC/RM abdomino-pélvica cada 2-3 años |
| Gammagrafía con MIBG 123I cada 2-3 años |
En nuestra serie, el 43% de los pacientes intervenidos eran hipertensos, se realizó por parte de medicina interna la determinación de catecolaminas en orina en 6 de ellos y todos los estudios fueron negativos. No pensamos que estén en relación con el tumor, al fin y al cabo. Según algunos estudios la prevalencia de hipertensión arterial en adultos se sitúa en el 35-40% y se acerca al 70% en población mayor de 60 años24,25
Algunos trabajos publicados4,5 hacen referencia a la búsqueda de tumores neuroendocrinos asociados o multicéntricos mediante TC o RM y gammagrafía, sin embargo no hacen referencia a la periodicidad ni a los estudios a familiares portadores de la mutación. Martínez-Aguilar et al. comentan que en pacientes asintomáticos portadores de la mutación se debe comenzar el estudio 10 años antes del caso más precoz diagnosticado en la familia con TC o RM de cuello, abdomen, tórax y pelvis cada 2 años y gammagrafía cada 2-3 años20.
Nosotros proponemos realizar un estudio genético a todos los pacientes diagnosticados de tumor del glomus carotídeo, y en los casos con mutación genética estudiar a los familiares de primer grado; ecodoppler anual de cuello a todos los pacientes y familiares portadores de mutación; y realizar determinación de catecolaminas en orina anualmente a todos los pacientes y a los familiares portadores de mutación. Los tumores del cuerpo carotídeo raramente son secretores de catecolaminas, por lo que una determinación positiva en orina nos obligaría a la búsqueda de otros focos de paragangliomas mediante TC o RM y gammagrafía. Debido a la mayor incidencia de multicentricidad y asociación a otros tumores en los casos familiares, sería aconsejable realizar TC o RM de abdomen y pelvis y gammagrafía de forma anual a los pacientes con mutación genética, y cada 2-3 años a los familiares asintomáticos portadores de la mutación y a los casos esporádicos.
Según los resultados obtenidos en nuestra serie podemos afirmar que la resección quirúrgica precoz es el tratamiento de elección de estos tumores y se asocia con una menor morbilidad (necesidad de reconstrucción vascular y lesión de pares craneales), y que por las peculiares características de los paragangliomas se nos plantea la necesidad de un estudio genético y un seguimiento sistematizado de los pacientes y familiares afectados.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Comunicación presentada en el «55 Congreso Nacional de la Sociedad Española de Angiología y Cirugía Vascular», Valencia, 18-20 de Junio de 2009.
