




Editado por: Sonia Roman
Más datosAdministration of carbon tetrachloride (CCl4), along with an hepatopathogenic diet, is widely employed as a chemical inducer to replicate human nonalcoholic steatohepatitis (NASH) in rodents; however, the role of the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome in this model remains unclear. We aimed to determine the relevance of NLRP3 inflammasome activation in the development of NASH induced by CCl4 along with an hepatopathogenic diet in male Wistar rats.
Materials and methodsAnimals were fed either a high fat, sucrose, and cholesterol diet (HFSCD) or a HFSCD plus intraperitoneal injections of low doses of CCl4 (400 mg/kg) once a week for 15 weeks. Liver steatosis, inflammation, fibrosis, and NLRP3 inflammasome activation were evaluated using biochemical, histological, ultrastructural, and immunofluorescence analyses, western blotting, and immunohistochemistry.
ResultsOur experimental model reproduced several aspects of the human NASH pathophysiology. NLRP3 inflammasome activation was induced by the combined effect of HFSCD plus CCl4 and significantly increased levels of both proinflammatory and profibrogenic cytokines and collagen deposition in the liver; thus, NASH severity was higher in the HFSCD+CCl4 group than that in the HFSCD group, to which CCl4 was not administered. Hepatic stellate cells, the most profibrogenic cells, were activated by HFSCD plus CCl4, as indicated by elevated levels of α-smooth muscle actin. Thus, activation of the NLRP3 inflammasome, triggered by low doses of CCl4, exacerbates the severity of NASH.
ConclusionsOur results indicate that NLRP3 inflammasome activation plays a key role and may be an important therapeutic target for NASH treatment.
Nonalcoholic steatohepatitis (NASH) is a major cause of global morbidity and can cause death by promoting cirrhosis and hepatocellular carcinoma (HCC) [1,2]. NASH is a chronic and progressive liver pathology caused by excessive caloric intake and a sedentary lifestyle that progresses from simple fatty liver to chronic liver inflammation [3]. An increased supply of lipids leads to elevated fatty acid oxidation, which triggers oxidative stress, induces cell death and apoptosis, and activates both inflammatory and fibrotic responses in the liver [4,5,6]. The prevalence of NASH and ineffective treatments have encouraged the development of experimental models to illustrate the pathophysiology and identify effective molecular targets that can interfere with disease progression [3]. Current diet-based animal models of NASH have limitations in recapitulating the late stages of the disease, such as fibrosis and/or HCC; therefore, researchers simultaneously administer special diets with chemical hepatotoxicants to reproduce the late stages of NASH in a short period of time [4,7,8].
Simple fatty liver, a reversible disease stage, progresses to NASH, a disease stage that may result in HCC and patient death. This outcome is, in part, due to the activation of a progressive proinflammatory process that triggers further alterations in the liver, such as fibrosis and cirrhosis; thus, the activation of proinflammatory signaling pathways plays a key role in NASH progression and represents an attractive therapeutic target.
Inflammasomes are large protein complexes that produce mature proinflammatory cytokines. These complexes detect intracellular danger signals; a well-described complex is the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) [9,10]. Upon inflammasome activation, a complex comprising NLRP3, the effector molecule procaspase 1, and the adapter protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) is formed. The formation of this complex leads to the activation of caspase 1 and the proteolytic cleavage and maturation of pro-interleukin (IL)-1β, yielding the biologically active form of IL-1β, which contributes to the inflammatory and fibrogenic responses during NASH progression [9,10]. However, the role of the NLRP3 inflammasome in NASH induced by an hepatopathogenic diet plus a chemical inducer has not been investigated. Therefore, we aimed to determine the involvement of the activation of the NLRP3 inflammasome in NASH progression induced by an experimental hepatopathogenic diet with simultaneous administration of carbon tetrachloride (CCl4) in rats.
2Materials and methods2.1Animal treatmentRats were obtained from the animal production and experimentation unit of the Center for Research and Advanced Studies of the National Polytechnic Institute (UPEAL–CINVESTAV–IPN; Mexico City, Mexico). NASH was induced in Wistar male rats by administering a high-fat, sucrose, and cholesterol diet (HFSCD), as previously reported [11]. Briefly, l.0% cholesterol, 0.5% sodium cholate, 5.0% butter, 30.0% sucrose, l0.0% casein, and 53.5% laboratory chow were mixed with 0.3% carboxymethyl cellulose (CMC), and the pellets were dried for a week at ± 40°C. Animals weighing 100–120 g were randomly divided into four groups (n = 8 per group). The control group was fed a regular diet (Labdiet® No. 5053, Indiana, USA) ad libitum for 15 weeks; the CCl4 group was intraperitoneally injected with CCl4 at 400 mg/kg once a week for 15 weeks; the HFSCD group was fed HFSCD ad libitum for 15 weeks; and animals in the HFSCD plus CCl4 group were fed HFSCD ad libitum for 15 weeks and were also subjected to an intraperitoneal injection of CCl4 once a week for 15 weeks. During the experimental procedure, body weight gain was recorded once per week. Animals had free access to water and were housed in polycarbonate cages under controlled conditions (21 ± 1°C, 50–60% relative humidity, and 12‐hour dark/light cycles). After the experimental procedure, the animals were anesthetized with ketamine and xylazine and euthanized by exsanguination. Blood was collected by cardiac puncture and centrifuged in tubes at 3,000 rpm (12,000 × g), and serum was separated. Furthermore, the liver was rapidly removed and stored at -75°C for further analyses.
2.2ReagentsCholesterol, cholate, and sucrose were purchased from Sigma-Aldrich (St. Louis, MO, USA). CCl4 was obtained from J.T. Backer (Xalostoc, Mexico, Mexico). Gloria® brand unsalted butter (Cremería Americana S.A. de C.V., Mexico City, Mexico) and Rennet Casein Powder 30 Mesh size (Irish Dairy Board Proteins and Ingredients, Dublin, Ireland) casein were used in this study.
2.3AntibodiesSupplementary Table 1 lists the antibodies against c-Jun N-terminal kinases (JNK), extracellular signal-regulated kinases (ERK), p38, p-JNK, p-ERK, p-p38, metalloproteinase (MMP)-13, Smad7, transforming growth factor-beta (TGF-β), smooth muscle alpha-actin (α-SMA), β-actin, desmin, connective tissue growth factor (CTGF), 4-hydroxynonenal (4-HNE), nuclear factor erythroid 2-related factor 2 (Nrf2), IL-1β, nuclear factor kappa B (NF-κB)-p65, tumor necrosis factor-alpha (TNF-α), toll-like receptor 4 (TLR4), sterol regulatory element-binding protein 1C (SREBP1C), peroxisome proliferator-activated receptor alpha (PPAR-α), NLRP3, ASC, and Caspase 1. The antibody dilutions for immunofluorescence (IF), immunohistochemistry (IHC), and western blot assays were 1:250, 1:250, and 1:500, respectively.
2.4Biochemical analysesActivity levels of alanine aminotransferase (ALT), gamma-glutamyl transpeptidase (γ-GTP), and alkaline phosphatase (AP) in the plasma were measured as previously described [12–14]. Liver glycogen content was quantified using the Anthrone method, and the absorbance was measured at 620 nm [15]. Liver-reduced glutathione (GSH) levels were quantified through the oxidation of GSH by the sulfhydryl reagent 5,5′-dithiol-bis (2-nitrobenzoic acid), which forms the yellow derivative 5-thio-2-nitrobenzoic acid, as previously described [16]. Lipid peroxidation (LPO) was measured in liver tissue by quantifying malondialdehyde (MDA) content using the thiobarbituric acid (TBA) procedure [17]. Triglyceride levels were determined in liver tissues, as previously described using a serum triglyceride determination kit (TR0100 Sigma–Aldrich®, St. Louis, MO, USA) [18]. Liver cholesterol levels were determined as previously described [19]. Protein concentration was measured using the Bradford method, and bovine serum albumin (BSA) was used as a standard [20]. Collagen content was assessed by determining the hydroxyproline content in hepatic pieces using Ehrlich's reagent (dimethylaminobenzaldehyde), as previously reported [21, 22].
2.5Histology determinationsHepatic samples were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS). Liver pieces were then embedded in paraffin, and 5 μm thick slides were obtained. Sections were prepared for IHC, hematoxylin and eosin (H&E), oil red O (ORO), or Masson trichrome staining. All stained slides were visualized under a light microscope (80i; Eclipse, Nikon®, Tokyo, Japan).
2.6Immunohistochemistry assaysLiver tissues were prepared for immunohistochemistry (IHC) staining using a standard immunoperoxidase protocol. Afterward, they were incubated overnight with primary antibodies against NF-κB (p65), TGF-β1, α-SMA, NLRP3, and Caspase 1 diluted in 3% fetal bovine serum. Then, the tissues were rinsed with 1X PBS five times for 5 min each. The stained samples were lightly counterstained with hematoxylin, dehydrated, resin-mounted, and dried for two days. Slides were visualized, and images were captured using a light microscope (80i, Eclipse, Nikon®, Tokyo, Japan). A positive label was detected as a brown gradient on the tissue, and the signal was quantified using ImageJ® software (NIH, MD, USA) [23].
2.7Transmission electron microscopyHepatic samples were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer for 1 h and then washed three times for 5–10 min each with the same buffer. Liver pieces were post-fixed for 1 h with 1% osmium tetroxide in 0.1 M sodium cacodylate, shaken lightly, and washed three times with 0.1 M sodium cacodylate for 5–10 min. The hepatic pieces were then dehydrated for 10 min with 70%, 80%, and 90% alcohol once and 100% alcohol twice, and for 20–30 min with propylene oxide twice. Afterward, the pieces were embedded in epoxy resin. Semi-thin sections were stained with toluidine blue. Thin sections (60–90 nm) were contrasted with uranyl acetate and lead citrate and examined using a JEOL 1400 transmission electron microscope (JEOL, Japan) [24].
2.8ImmunofluorescenceFor double-labeling immunofluorescence (IF) of NLRP3/caspase 1 and NLRP3/α-SMA, liver sections were permeabilized with 0.2% PBS-Triton for 20 min at room temperature. Thereafter, the samples were treated with 5% BSA for 15 min at room temperature and incubated overnight at 4°C with primary antibodies (Supplementary Table 1). The hepatic pieces were then washed with 0.2% PBS-triton and incubated for 2 h with secondary antibodies (Supplementary Table 1) diluted in 0.2% PBS-triton at room temperature. Control tissue samples were processed in the absence of primary antibodies. All hepatic samples were co-incubated with 4′, 6′-diamidino-2 phenylindole (1:1,000) (Invitrogen®, MA, USA) in 0.2% PBS-triton for 30 min at room temperature. Finally, the tissue samples were washed, dried, and coverslipped with the mounting medium VectaShield-H-1000 (Vector Laboratories®, CA, USA) [25].
2.9Western blotting analysisLiver tissue was homogenized in lysis buffer (Sigma-Aldrich®, MO, USA) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich®, MO, USA) and then centrifuged at 12,000 rpm (13,200 × g) for 20 min at 4°C. The supernatant was recovered, and the protein concentration was measured using the bicinchoninic acid method [26] (Pierce BCA Protein Assay, Cat. no. 23223; Thermo Fischer Scientific, NY, USA). Equivalent amounts of protein were analyzed using 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a 0.45 µm Immuno-Blot PVDF membrane (BIO-RAD®, CA, USA). The membranes were blocked using 5% BSA (Sigma-Aldrich®, MO, USA) in Tris-buffered saline with 0.1% Tween-20 (TBST) for 2 h at room temperature. Membranes were then incubated with primary antibodies (Supplementary Table 1) overnight at 4°C, washed, and incubated with secondary antibodies for 2 h at room temperature. The β-actin protein level was used as an internal control, and results were expressed as a ratio compared to controls. For signal detection, membranes were bathed in luminol reagent (Santa Cruz Biotechnology®, CA, USA). Photographic plates (catalog number 822526, Kodak®, NY, USA) were digitized, and band intensities were quantified by densitometry using ImageJ® software (NIH, MD, USA) [23].
2.10ZymographyProteolytic activity was evaluated on gelatin-substrate gels, as previously described [24]. The protein content was determined by the bicinchoninic acid method [26]. Images were digitized, and band intensities were quantified using ImageJ® software (NIH, MD, USA) [23].
2.11Statistical analysesAll data are shown as mean ± standard error of the mean (SEM). Multiple comparisons were performed using the GraphPad Prism software (version 7.0; CA, USA). Data were analyzed using one-way ANOVA followed by Tukey's test for multiple comparisons. Differences were considered to be statistically significant at P ≤ 0.05.
2.12Ethical statementThe investigation was performed in accordance with the guidelines of the Institution and the official Mexican regulations (NOM-062-ZOO-1999) with technical specifications for the production, care, and handling of laboratory animals according to the Guide for the Care and Use of Laboratory Animals (NRC, 2011). The Ethical Committee of the Animal Lab Facility of the Cinvestav-IPN approved the protocol (numbers 281-19).
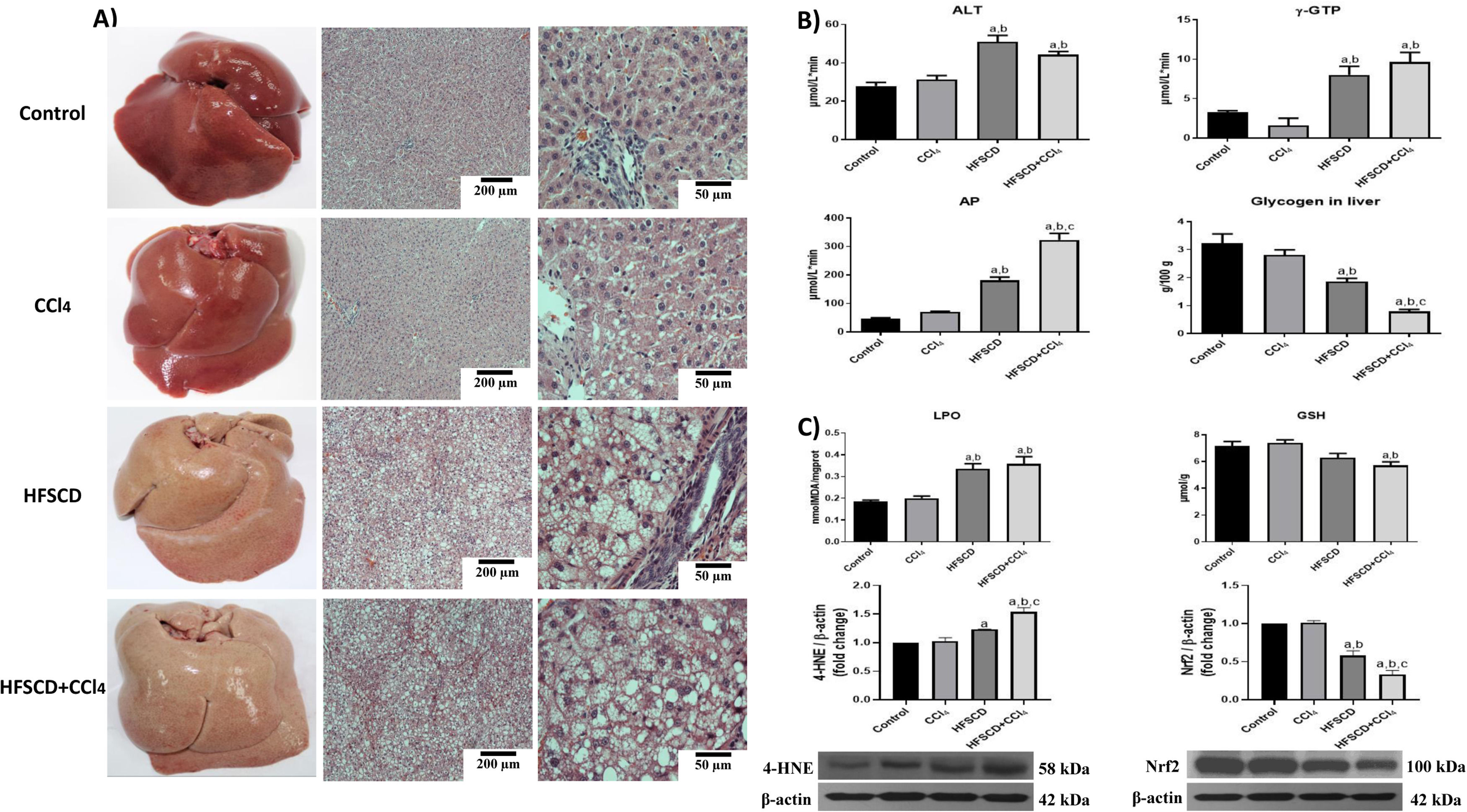
3Results3.1HFSCD and HFSCD+CCl4 protocols increase liver damage and oxidative stress markers in ratsMacroscopic examination of the livers of both the control and CCl4 groups revealed a typical, smooth, and shiny appearance (Fig. 1A). In contrast, livers from the HFSCD and HFSCD+CCl4 groups were extremely pale, whitish, and enlarged. Microscopic examination of H&E-stained tissues from the control and CCl4 groups showed normal polyhedral hepatocytes with central nuclei and eosinophilic cytoplasm (Fig. 1A). In contrast, both the HFSCD and HFSCD+CCl4 groups showed marked disruption of the hepatic parenchyma, ballooning degeneration of hepatocytes, loss of cytoplasmic eosin, eccentric nuclei, diffuse microvesicular and macrovesicular steatosis, portal inflammation, foci of lobular inflammation, and necrosis (Fig. 1A); in addition, bands of connective tissue between hepatocytes were observed in the HFSCD+CCl4 group. The structural changes in the HFSCD and HFSCD+CCl4 groups were accompanied by increased levels of liver injury markers, such as ALT, γ-GTP, and AP serum activities (Fig. 1B). Some liver damage parameters, such as AP activity and hepatic glycogen content, were higher in the HFSCD+CCl4 group than those in the HFSCD group (Fig. 1B).
 Macroscopic and microscopic anatomy of the liver. Hematoxylin and eosin stained sections of livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 200 μm and 50 μm. B) General markers of liver damage. Serum ALT, γ-GTP, and AP activities, and hepatic glycogen content in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean of experiments performed in duplicate ± SEM (n = 8). C) Oxidative stress markers. Degree of LPO and content of GSH in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean ± SEM (n = 8). Western blotting of 4-HNE and Nrf2 proteins in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean ± SEM of duplicate experiments (n = 3), and β-actin was used as a loading control. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, ALT: alanine aminotransferase, γ-GTP: gamma-glutamyl transpeptidase, AP: alkaline phosphatase, SEM: standard error of the mean, LPO: lipid peroxidation, GSH: glutathione, 4-HNE: 4-hydroxynonenal, Nrf2: nuclear factor erythroid 2-related factor 2.")
Macroscopic and microscopic anatomy of the liver, general markers of liver damage, and oxidative stress parameters of rats fed HFSCD and HFSCD+CCl4. A) Macroscopic and microscopic anatomy of the liver. Hematoxylin and eosin stained sections of livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 200 μm and 50 μm. B) General markers of liver damage. Serum ALT, γ-GTP, and AP activities, and hepatic glycogen content in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean of experiments performed in duplicate ± SEM (n = 8). C) Oxidative stress markers. Degree of LPO and content of GSH in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean ± SEM (n = 8). Western blotting of 4-HNE and Nrf2 proteins in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean ± SEM of duplicate experiments (n = 3), and β-actin was used as a loading control. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, ALT: alanine aminotransferase, γ-GTP: gamma-glutamyl transpeptidase, AP: alkaline phosphatase, SEM: standard error of the mean, LPO: lipid peroxidation, GSH: glutathione, 4-HNE: 4-hydroxynonenal, Nrf2: nuclear factor erythroid 2-related factor 2.
Fig. 1C shows increased LPO in both the HFSCD and HFSCD+CCl4 groups as MDA levels were significantly elevated. Interestingly, liver GSH concentration, one of the main endogenous antioxidants, was significantly decreased only in the HFSCD+CCl4 group. In the livers of animals subjected to HFSCD and HFSCD+CCl4 protocols, the levels of 4-HNE, an LPO product and marker of oxidative stress, were significantly increased, and this level was higher in the HFSCD+CCl4 group than that in the HFSCD group. The HFSCD and HFSCD+CCl4 protocols reduced the levels of Nrf2; this effect was stronger in the HFSCD+CCl4 group than that in the HFSCD group. Unlike the animals subjected to HFSCD and HFSCD+CCl4 protocols, both liver damage and oxidative stress markers were not modified by the CCl4 protocol alone (Fig. 1).
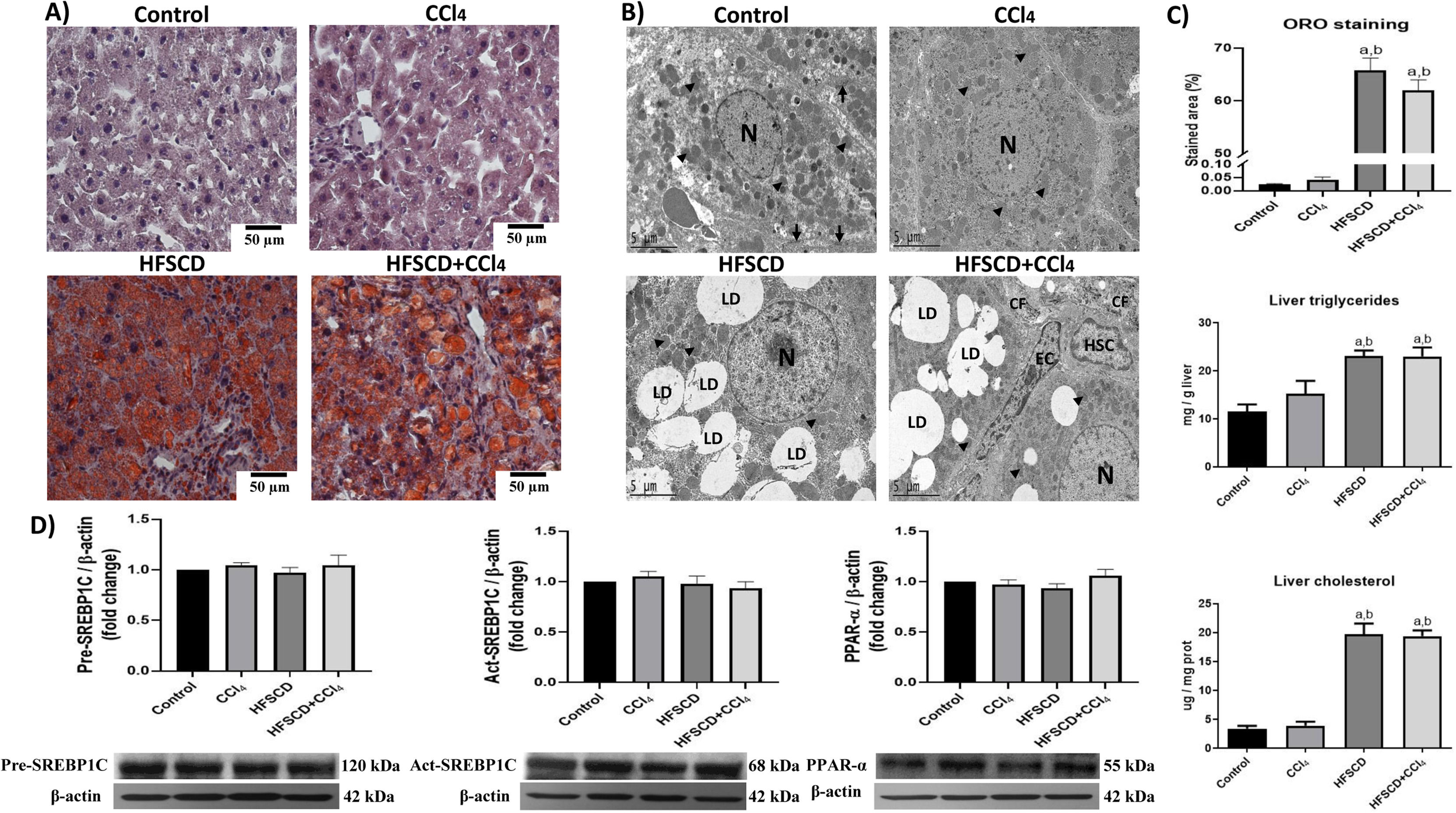
3.2HFSCD and HFSCD+CCl4 protocols induce steatosis and modify the liver ultrastructureHepatic steatosis was histochemically determined by Oil red O (ORO) staining (Fig. 2A). Animals subjected to the HFSCD and HFSCD+CCl4 protocols exhibited marked liver steatosis, which was corroborated by measuring the content of triglyceride and cholesterol in the liver (Fig. 2C). SREBP-1C and PPAR-α protein levels (Fig. 2D)—associated with de novo lipogenesis and free fatty acid oxidation, respectively—were evaluated by western blot analysis. No significant differences were observed between the experimental groups. However, the ultrastructure of the liver was disrupted in animals subjected to HFSCD and HFSCD+CCl4 protocols, showing an abundant accumulation of lipid droplets and partial mitochondrial disarrangement (Fig. 2B). Importantly, images from the livers of animals of the HFSCD+CCl4 group showed activated HSCs, which are the main producers of the extracellular matrix, as well as areas of collagen deposition, whereas animals treated with CCl4 alone did not show any significant alterations (Fig. 2).
 Oil red O (ORO) staining of livers from animals in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 50 μm. B) Transmission electron microscopy (TEM) of samples from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 5 μm; (black arrow) tight junctions, (arrowhead) mitochondria, (CF) collagen fibers, (HSC) hepatic stellate cell, (LD) lipid drop, (N) nucleus, and (EC) endothelial cell. C) Percentage of the ORO staining area. Bars represent the mean value ± SEM (n = 4) of liver triglycerides and liver cholesterol. Bars represent the mean value ± SEM (n = 5). D) Western blot of precursor-sterol regulatory element-binding protein 1C (pre-SREBP1C), active-SREBP1C (act-SREBP1C), and PPAR-α proteins from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean ± SEM (n = 3) of duplicate experiments, and β-actin was used as a control. a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group. HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, SEM: standard error of the mean, PPAR-α: peroxisome proliferator-activated receptor alpha.")
Steatosis markers and the liver ultrastructure of rats subjected to the HFSCD and HFSCD+CCl4 protocols. A) Oil red O (ORO) staining of livers from animals in the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 50 μm. B) Transmission electron microscopy (TEM) of samples from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 5 μm; (black arrow) tight junctions, (arrowhead) mitochondria, (CF) collagen fibers, (HSC) hepatic stellate cell, (LD) lipid drop, (N) nucleus, and (EC) endothelial cell. C) Percentage of the ORO staining area. Bars represent the mean value ± SEM (n = 4) of liver triglycerides and liver cholesterol. Bars represent the mean value ± SEM (n = 5). D) Western blot of precursor-sterol regulatory element-binding protein 1C (pre-SREBP1C), active-SREBP1C (act-SREBP1C), and PPAR-α proteins from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Bars represent the mean ± SEM (n = 3) of duplicate experiments, and β-actin was used as a control. a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group. HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, SEM: standard error of the mean, PPAR-α: peroxisome proliferator-activated receptor alpha.
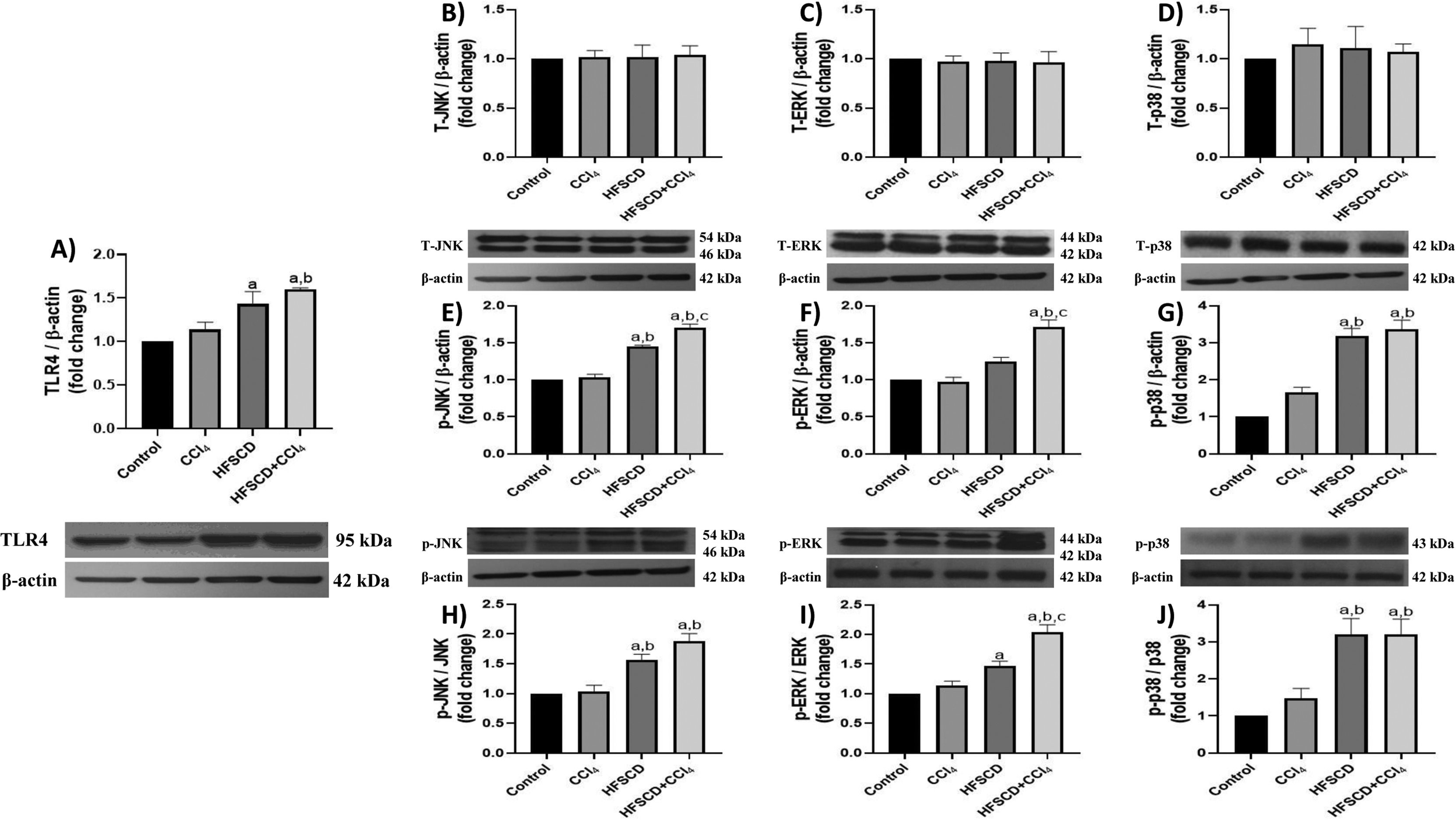
TLR4 levels were significantly increased in the HFSCD and HFSCD+CCl4 groups compared to those in controls (Fig. 3A). Total JNK, ERK, and p38 protein levels were not affected by any of the experimental groups (Fig. 3B-D); however, the phosphorylated forms of these proteins significantly increased in the livers of rats from the HFSCD and HFSCD+CCl4 groups (Fig. 3E-G). Additionally, the phosphorylated forms of ERK and JNK proteins were significantly higher in the HFSCD+CCl4 group than in the HFSCD group (Fig. 3E-F). The levels of these proteins in livers from the CCl4 group did not show any changes.
, JNK (B), ERK (C), p38 (D), p-JNK (E), p-ERK (F), and p-p38 (G) in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups were determined by western blot analysis. Bars represent the mean ± SEM (n = 3) of duplicate experiments, and β-actin was used as the loading control. p-JNK/JNK (H), p-ERK/ERK (I), and p-p38/p38 (J) ratios. Values are presented as the fold change of the optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. TLR4: toll-like receptor 4, MAPK: mitogen-activated protein kinase, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, JNK: c-Jun N-terminal kinases, ERK: extracellular signal-regulated kinases, SEM: standard error of the mean.")
TLR4/MAPK pathway in rats from the HFSCD and HFSCD+CCl4 groups. The protein levels of TLR4 (A), JNK (B), ERK (C), p38 (D), p-JNK (E), p-ERK (F), and p-p38 (G) in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups were determined by western blot analysis. Bars represent the mean ± SEM (n = 3) of duplicate experiments, and β-actin was used as the loading control. p-JNK/JNK (H), p-ERK/ERK (I), and p-p38/p38 (J) ratios. Values are presented as the fold change of the optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. TLR4: toll-like receptor 4, MAPK: mitogen-activated protein kinase, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, JNK: c-Jun N-terminal kinases, ERK: extracellular signal-regulated kinases, SEM: standard error of the mean.
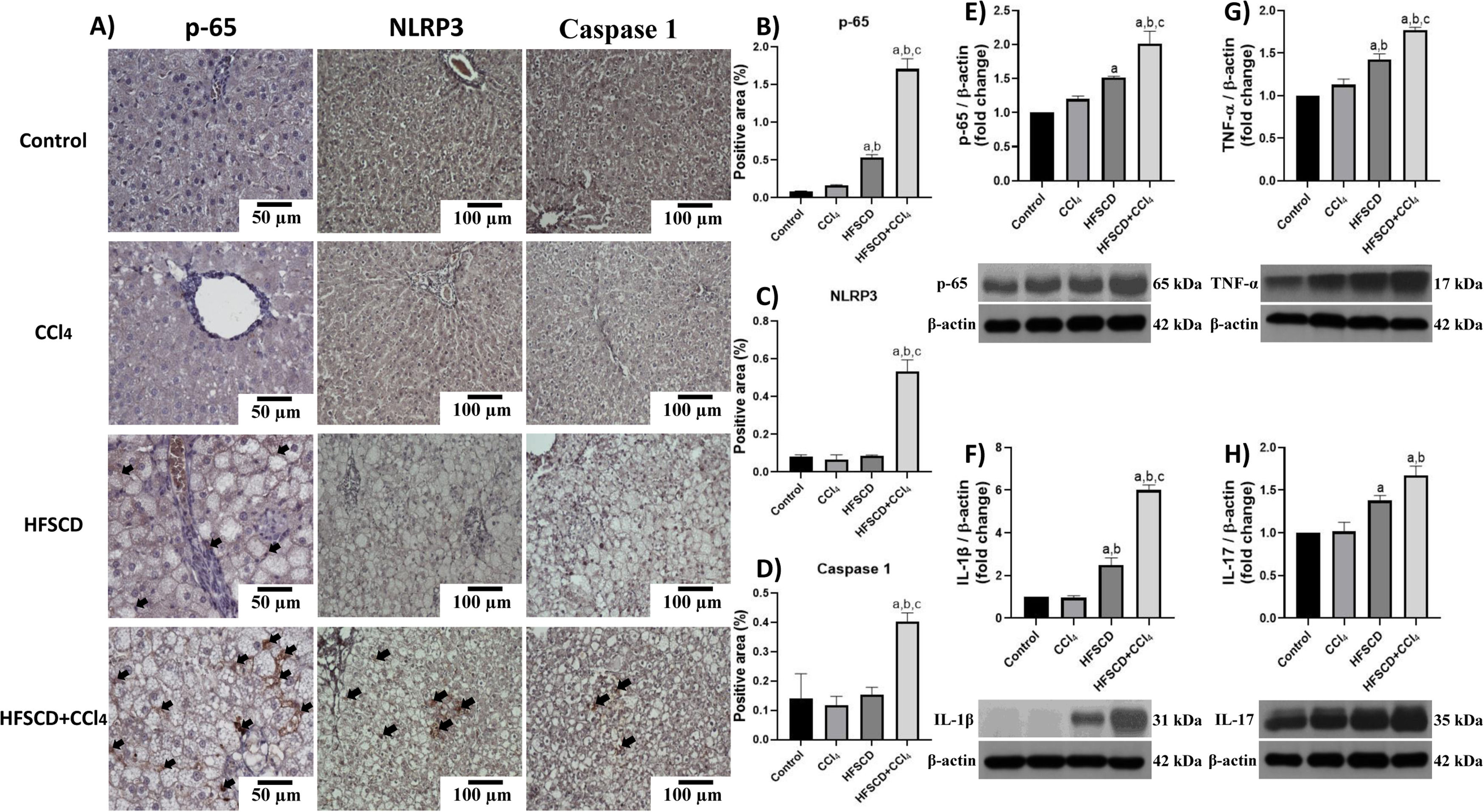
Fig. 4A shows p-65, NLRP3, and caspase 1-specific antigen detection by IHC analysis. The percentage of p-65-, NLRP3-, and caspase 1-positive areas are shown in Fig. 4B, C, and D, respectively. While the HFSCD+CCl4 protocol significantly induced the production of all three proteins, only p65 production was induced by HFSCD alone (Fig. 4B-D). Western blot analysis of p65 confirmed this finding (Fig. 4E). We also measured the levels of IL-1β, TNF-α, and IL-17, which are upregulated by the NF-κB signaling pathway. Interestingly, livers from the HFSCD and HFSCD+CCl4 groups showed a significant increase in these proinflammatory cytokines (Fig. 4F-H) as compared to livers of the control group. In addition, the levels of p65, IL-1β, TNF-α, and IL-17 were higher in the HFSCD+CCl4 group than in the HFSCD group (Fig. 4E-H).
, proinflammatory cytokines, and NLRP3 in livers from the HFSCD and HFSCD+CCl4 groups. A) Representative IHC images of p65, NLRP3, and caspase 1 in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 50 μm and 100 μm. The percentages of the positive label for p65 (B), NLRP3 (C), and caspase 1 (D) were obtained from IHC slices (n = 4). The positivity label is indicated by arrows. Levels of p65 (E), interleukin (IL)-1β (F), TNF-α (G), and IL-17 (H) proteins were detected by western blot (n =3). β-Actin was used as the loading control. Values are presented as the fold change of optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. NF-κB: nuclear factor kappa B, NLRP3: nucleotide-binding domain leucine-rich-containing family, pyrin domain-containing-3, IHC: immunohistochemistry, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, TNF-α: tumor necrosis factor-alpha, SEM: standard error of the mean.")
Level of NF-κB (p65), proinflammatory cytokines, and NLRP3 in livers from the HFSCD and HFSCD+CCl4 groups. A) Representative IHC images of p65, NLRP3, and caspase 1 in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bar = 50 μm and 100 μm. The percentages of the positive label for p65 (B), NLRP3 (C), and caspase 1 (D) were obtained from IHC slices (n = 4). The positivity label is indicated by arrows. Levels of p65 (E), interleukin (IL)-1β (F), TNF-α (G), and IL-17 (H) proteins were detected by western blot (n =3). β-Actin was used as the loading control. Values are presented as the fold change of optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. NF-κB: nuclear factor kappa B, NLRP3: nucleotide-binding domain leucine-rich-containing family, pyrin domain-containing-3, IHC: immunohistochemistry, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, TNF-α: tumor necrosis factor-alpha, SEM: standard error of the mean.
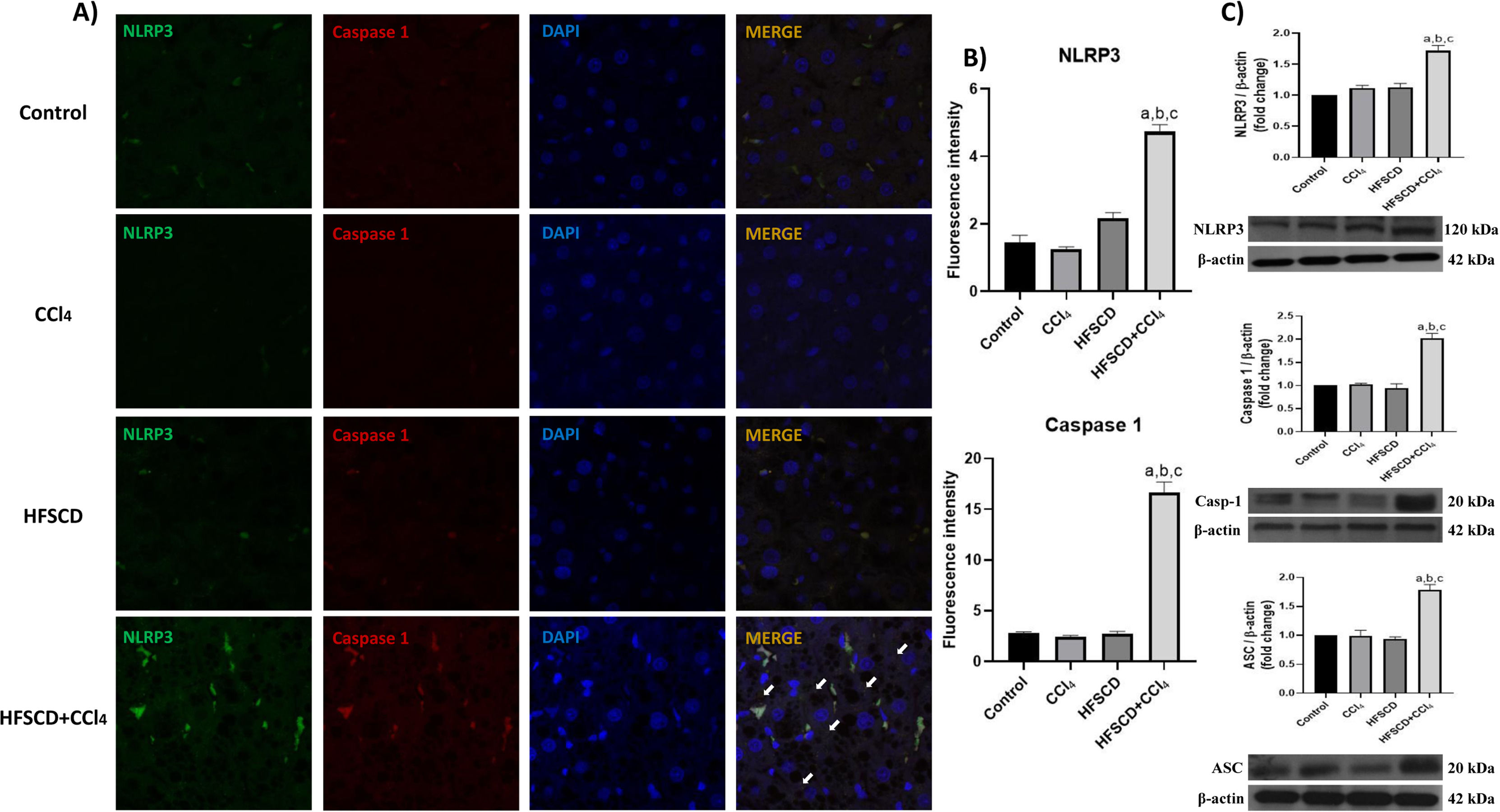
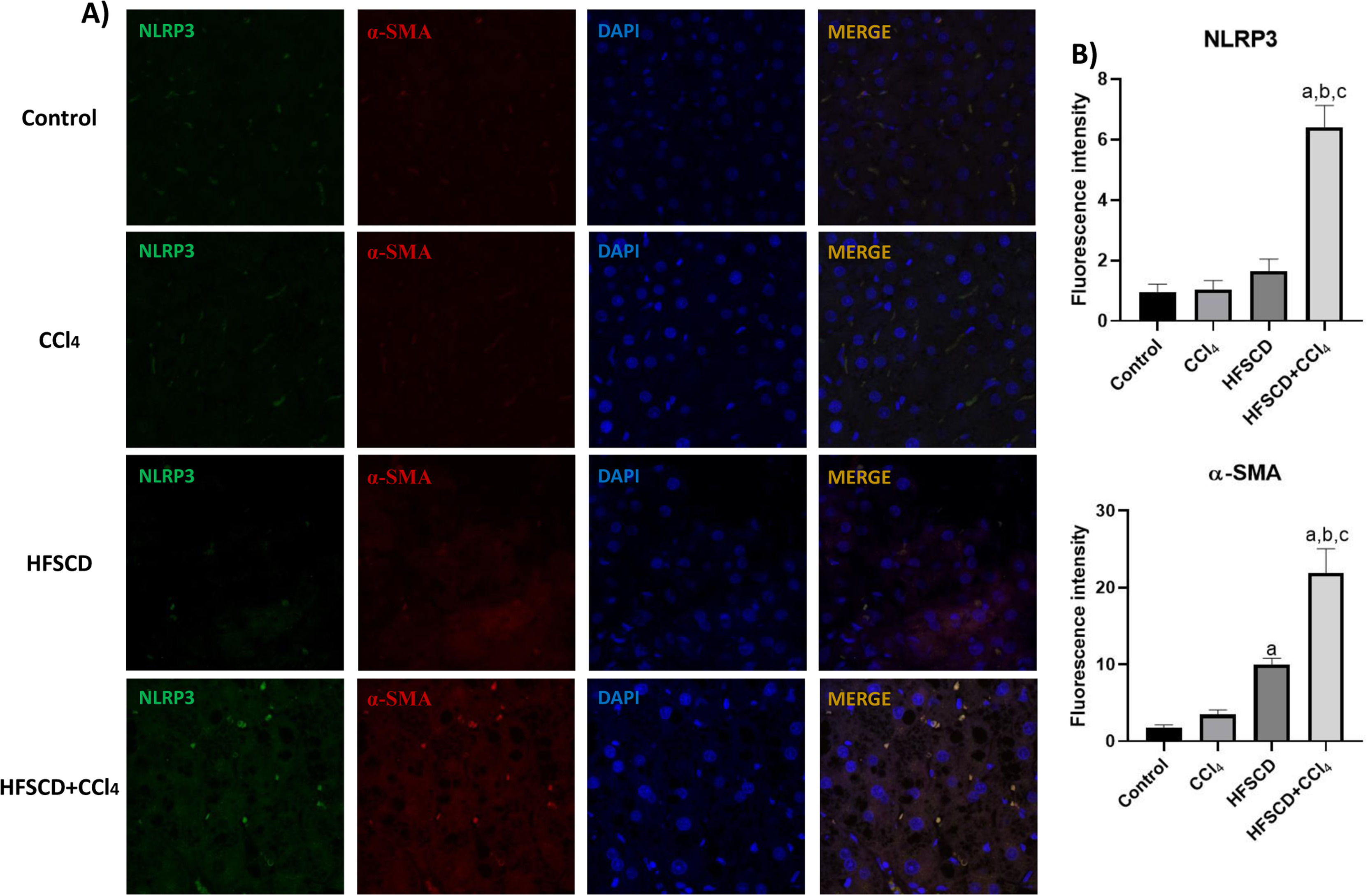
The colocalization of NLRP3 and caspase 1 was investigated using IF analysis (Fig. 5A). The analysis revealed that livers from the control, CCl4, and HFSCD groups did not show the formation of an inflammasome complex; however, in livers from the HFSCD+CCl4 group, markers of inflammasome-associated proteins, namely NLRP3 and Caspase 1, were observed and their labels were colocalized, indicating that they were interacting in the active form of NLRP3 inflammasome (Fig. 5A). The signal quantification of these proteins is shown in Fig. 5B. Western blot analysis of these proteins confirmed this phenomenon (Fig. 5C), and the level of ASC protein was also determined. Fig. 5C shows that the level of this protein was significantly increased only in the livers of the HFSCD+CCl4 group. In addition, we determined the colocalization of NLRP3 and α-SMA proteins using IF analysis (Fig. 6A). Quantification of the fluorescence intensity showed a correlation between the NLRP3 and α-SMA proteins in the livers of the HFSCD+CCl4 group.
 IF labeling of NLRP3/caspase 1 in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. White arrows indicate matches between the procaspase 1 and NLRP3 inflammasome labels. B) Fluorescence intensity of NLRP3 and caspase 1 was quantified from IF slices (n = 3). C) Levels of NLRP3, ASC, and caspase 1 proteins in liver tissues were detected by western blotting (n =3). β-Actin was used as the loading control. Values are presented as the fold change of the optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. NLRP3: nucleotide-binding domain leucine-rich-containing family, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, IF: immunofluorescence, ASC: adapter protein apoptosis-associated speck-like protein containing a caspase recruitment domain, SEM: standard error of the mean.")
Double labeling of NLRP3 and caspase 1 in livers from the HFSCD and HFSCD+CCl4 groups. A) IF labeling of NLRP3/caspase 1 in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. White arrows indicate matches between the procaspase 1 and NLRP3 inflammasome labels. B) Fluorescence intensity of NLRP3 and caspase 1 was quantified from IF slices (n = 3). C) Levels of NLRP3, ASC, and caspase 1 proteins in liver tissues were detected by western blotting (n =3). β-Actin was used as the loading control. Values are presented as the fold change of the optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. NLRP3: nucleotide-binding domain leucine-rich-containing family, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, IF: immunofluorescence, ASC: adapter protein apoptosis-associated speck-like protein containing a caspase recruitment domain, SEM: standard error of the mean.
 IF labeling of NLRP3/α-SMA in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. B) Fluorescence intensity of NLRP3 and α-SMA was quantified from IF slices (n = 3). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. NLRP3: nucleotide-binding domain leucine-rich-containing family, α-SMA: smooth muscle alpha-actin, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, IF: immunofluorescence, SEM: standard error of the mean.")
Double labeling of NLRP3 and α-SMA in livers from the HFSCD and HFSCD+CCl4 groups. A) IF labeling of NLRP3/α-SMA in livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. B) Fluorescence intensity of NLRP3 and α-SMA was quantified from IF slices (n = 3). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. NLRP3: nucleotide-binding domain leucine-rich-containing family, α-SMA: smooth muscle alpha-actin, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, IF: immunofluorescence, SEM: standard error of the mean.
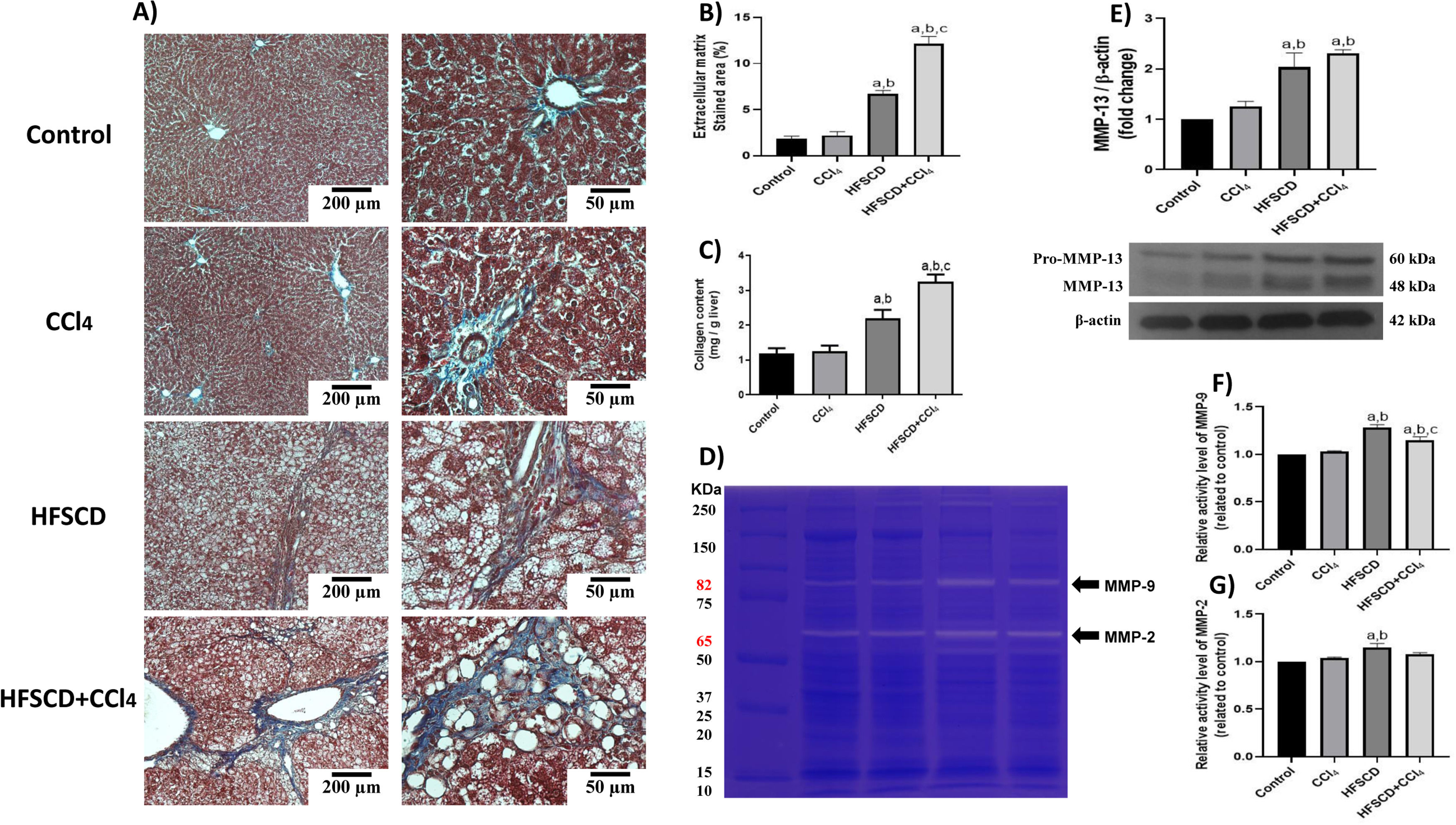
Scar tissue accumulation within the liver parenchyma was visualized using Masson´s trichrome staining, and collagen was biochemically quantified (Fig. 7A-C). Importantly, both the HFSCD and HFSCD+CCl4 protocols induced elevation in collagen levels; however, livers from the HFSCD+CCl4 group exhibited higher collagen levels than those from the HFSCD group, and CCl4 alone did not induce any collagen deposition. Moreover, ECM degradation was assessed by determining MMP-2 and MMP-9 activity as well as the protein level of MMP-13 (Fig. 7D-G). Gelatinase activity was elevated in hepatic tissues from the HFSCD group, but MMP-2 activity was not significantly increased in the livers of the HFSCD+CCl4 group. The western blot analysis results revealed that the MMP-13 protein levels were increased in the livers of the HFSCD and HFSCD+CCl4 groups (Fig. 7E).
 Masson")
Fibrosis and profibrogenic mediators and MMPs in livers from the HFSCD and HFSCD+CCl4 groups. A) Masson's trichrome staining of livers from the control, CCl4, HFSCD, and HFSCD+CCl4 groups. Scale bars = 200 μm and 50 μm. Percentages of collagen areas (B) (n = 4). The collagen level was determined by measuring the levels of hydroxyproline in the liver (C) (n = 6). The activities of MMP-9 (F) and MMP-2 (G) were determined by zymography (D) (n = 5). Bars represent the mean ± SEM. Values are presented as fold change of the optical density normalized to values of the control group (control = 1). Protein levels of MMP-13 (E) from livers were examined by western blot analysis (n = 3), and β-actin was used as the loading control. Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. MMP: metalloproteinase, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, SEM: standard error of the mean.
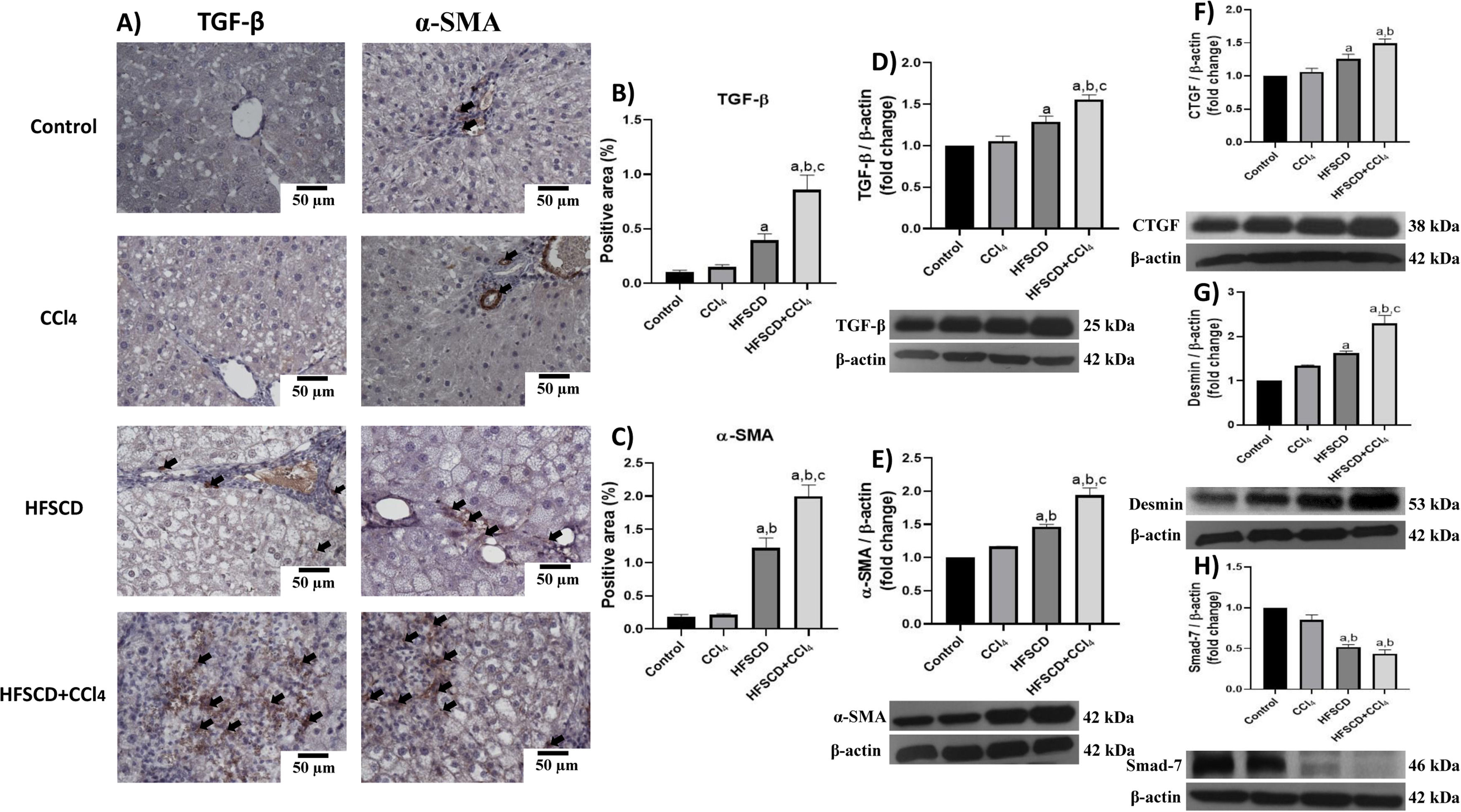
We then evaluated the effects of the experimental protocols on fibrogenesis-associated proteins. Both IHC and Western blot analyses showed that the HFSCD and HFSCD+CCl4 groups exhibited significantly increased levels of TGF-β and α-SMA proteins as compared to the control group (Fig. 8A-E). Notably, the co-administration of CCl4 with HFSCD induced higher levels of TGF-β and α-SMA proteins than HFSCD alone. Moreover, the levels of CTGF protein, as well as that of desmin, which is a specific marker of HSCs, were significantly augmented in both the HFSCD and HFSCD+CCl4 groups; however, the latter group exhibited a higher increment (Fig. 8F and G). In addition, the level of Smad7, an antifibrotic Smad protein, was significantly decreased in the livers of the HFSCD and HFSCD+CCl4 groups (Fig. 8H), and CCl4 alone did not affect the levels of these parameters. Taken together, these results indicate that CCl4 exacerbates both steatohepatitis and fibrosis induced by HFSCD.
 Representative IHC images of TGF-β and α-SMA levels in livers from the control, CCl4, HFSCD and HFSCD+CCl4 groups. Scale bar = 50 μm. A positive area of TGF-β (B) and α-SMA is shown in the histogram (C) (n = 4). The protein levels of TGF-β (D), α-SMA (E), CTGF (F), desmin (G), and Smad-7 (H) in livers from the control, CCl4, HFSCD and HFSCD+CCl4 groups were determined by Western blot (n = 3), and β-actin was used as the loading control. Values are presented as the fold change of the optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. TGF-β: transforming growth factor-beta, α-SMA: smooth muscle alpha-actin, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, IHC: immunohistochemistry, CTGF: connective tissue growth factor, SEM: standard error of the mean.")
Levels of TGF-β, α-SMA, and profibrogenic mediators in livers from HFSCD and HFSCD+CCl4 groups. A) Representative IHC images of TGF-β and α-SMA levels in livers from the control, CCl4, HFSCD and HFSCD+CCl4 groups. Scale bar = 50 μm. A positive area of TGF-β (B) and α-SMA is shown in the histogram (C) (n = 4). The protein levels of TGF-β (D), α-SMA (E), CTGF (F), desmin (G), and Smad-7 (H) in livers from the control, CCl4, HFSCD and HFSCD+CCl4 groups were determined by Western blot (n = 3), and β-actin was used as the loading control. Values are presented as the fold change of the optical density normalized to values of the control group (control = 1). Bars represent the mean ± SEM. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the CCl4 group; (c) P < 0.05 compared with the HFSCD group. TGF-β: transforming growth factor-beta, α-SMA: smooth muscle alpha-actin, HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, IHC: immunohistochemistry, CTGF: connective tissue growth factor, SEM: standard error of the mean.
The interest in uncovering the pathogenesis of NAFLD and the more severe stage, NASH, has significantly increased in recent decades owing to their considerable impacts on health worldwide. NASH is associated with inflammation and pericellular fibrosis, which can lead to cirrhosis and HCC. Animal models of NASH have significantly contributed to mimicking human diseases by illustrating the cellular and molecular mechanisms implicated in its progression. Moreover, their contribution goes beyond the mere description of NASH pathogenesis because they may be useful in identifying therapeutic targets and biomarkers of liver damage for early diagnosis and treatment. In most previously proposed models, fatty liver was induced by high-sugar and high-fat diets; however, these methods have limitations in recapitulating NASH, fibrosis, cirrhosis, and HCC. To overcome these limitations, some researchers have developed alternative models in which, concomitantly with diets, chemical inducers, such as CCl4, are administered [3,7]; however, the role of CCl4 in NASH models has not been investigated in depth. Tsuchida et al. [7] developed a NASH model in which animals were fed a diet containing high amounts of fructose, fat, and cholesterol and were administered a weekly injection of low doses of CCl4, a well-known hepatotoxic agent, to potentiate the effect of diet. The dose used in this research of 400mg/kg of body weight once per week is much lower than the dose that is usually given for fibrosis induction with CCl4 alone in rats.
Inflammation is considered a fundamental step in the progression of fatty liver to NASH and plays a key role in fibrogenesis [27,28], Here, the role of the NLRP3 inflammasome and NF-κB signaling pathways were investigated as targets of CCl4 for the induction of NASH in an animal model subjected to an hepatopathogenic diet. Inflammasome activation is triggered by a range of substances that emerge during infections, tissue damage, or metabolic imbalances [29]. In the context of NASH, cholesterol crystals, ROS, and fatty acids may activate inflammasomes [30]. Activation of the NLRP3 inflammasome plays a key role in the progression of human NASH [31–33]; therefore, we investigated whether this inflammatory signaling pathway is targeted as an aberrant action mechanism in a NASH model induced by the concomitant effect of HFSCD and CCl4.
Human NASH is histologically characterized by steatosis, inflammatory infiltrates, and fibrosis, which may or may not be present [4,34]. Animals fed HFSCD alone or HFSCD plus CCl4 were found to reproduce these characteristics. We also found increased activity of the liver damage enzymes ALT, γ-GTP, and AP, a phenomenon that frequently occurs in patients with both NAFLD and NASH [35,36].
In this study, the hepatic fat content was quantified utilizing the area stained by ORO and by the quantification of hepatic cholesterol and triglycerides. We observed, by quantifying the ORO staining area, a dramatic and significant increase in hepatic fat deposition in the HFSCD and HFSCD plus CCl4 groups. Because the accumulation of fat within the organ is progressive, it would be expected that the hepatic fat content would increase depending on the treatment period. We did not find significant differences in hepatic steatosis between animals subjected to HFSCD alone and those subjected to HFSCD+CCl4, suggesting that CCl4 administration does not exacerbate lipid deposition. In this study, CCl4 administration did not modify the proteins levels of SREBP1C or PPAR-α, which are associated with de novo lipogenesis and free fatty acid oxidation, respectively, in different experimental models. These results are in concordance with previous data obtained from patients with different NASH degrees [37], in which PPAR-α levels were not modified in any NASH stage, and SREBP-1C levels were only increased during the simple steatosis stage, but remained at a physiological level in mild NASH, similar to our current findings.
An important factor contributing to human NASH is oxidative stress induced by the increasing supply of lipids to the liver, which exacerbates the oxidation of fatty acids [4,5,6]. Animals subjected to the HFSCD and HFSCD+CCl4 protocols exhibited increased oxidative parameters; for example, 4-HNE, an intermediate product of lipid peroxidation [38], was significantly elevated. Interestingly, the protein levels of the master regulator of the antioxidant response in cells, Nrf2 [39], were decreased in the HFSCD+CCl4 group as compared with those in the HFSCD group, which strongly suggests that CCl4 may enhance reactive oxygen species (ROS) production in HFSCD-fed rats but not by itself. A plausible explanation for this phenomenon is that HFSCD sensitizes liver cells, and as a result, the oxidative damage induced by CCl4 is increased.
ROS attack proteins, lipids, and DNA, leading to defective organelle function; they may cause cell death and activate proinflammatory signaling pathways, which are responsible for the progression from simple fatty liver to reversible liver alteration to harmful NASH [4,5,6]. Indeed, rats fed HFSCD exhibited increased TLR4 and MAPK protein levels, which triggered NF-κB activation, the master regulator of inflammation [40]. Interestingly, weekly administration of low CCl4 doses to HFSCD-fed rats elevated the protein levels of p-JNK, p-ERK, and NF-κB levels, indicating that the concomitant effect of HFSCD and CCl4 exacerbates liver inflammation. These results clearly show that CCl4 administration to rats fed an hepatopathogenic diet potentiates experimental NASH progression and recapitulates the disease as it occurs in humans. The NF-κB signaling pathway upregulates NLRP3, pro-IL-1β, and proinflammatory cytokines such as IL-17 and TNF-α. NLRP3 can also be activated by multiple danger signals, including ROS, cholesterol crystals, and fatty acids [41]. Surprisingly, we found that HFSCD alone was not sufficient to induce the activation of the NLRP3 inflammasome; however, the concomitant weekly administration of low CCl4 doses to HFSCD-fed rats strongly triggered activation of this powerful proinflammatory signaling pathway. Therefore, our results indicate that HFSCD induces inflammation through activation of the NF-κB signaling pathway; however, when low CCl4 doses are concomitantly administered, the NRLP3 inflammasome pathway is activated to further potentiate inflammation and fibrosis. In addition, a positive relationship between increased NLRP3 inflammasome activation and levels of α-SMA—a protein associated with HSC and myofibroblast activation—was observed. Moreover, desmin, a selective HSC marker, was also correlated with NLRP3 inflammasome activation. HSCs are considered the main producers of extracellular matrix proteins in the liver [42]. Smad7 inhibits the TGF-β fibrogenic signaling pathway [43]. Notably, we observed that Smad7 protein levels were decreased in the livers of the HFSCD and HFSCD+CCl4 groups. Together, this evidence indicates that fibrogenic pathways are also concomitantly stimulated by HFSCD plus CCl4 treatment to persistently maintain fibrogenesis. Importantly, weekly low-dose CCl4 administration to rats did not induce fibrosis; however, when it was administered to the rats fed with the hepatopathogenic diet, fibrosis increased significantly, suggesting that CCl4 is acting as a chemical inductor rather as a profibrogenic agent by itself at this low dose. Finally, our evidence provides novel data regarding the similarities between the progression of an experimental model and human NASH (Fig. 9).
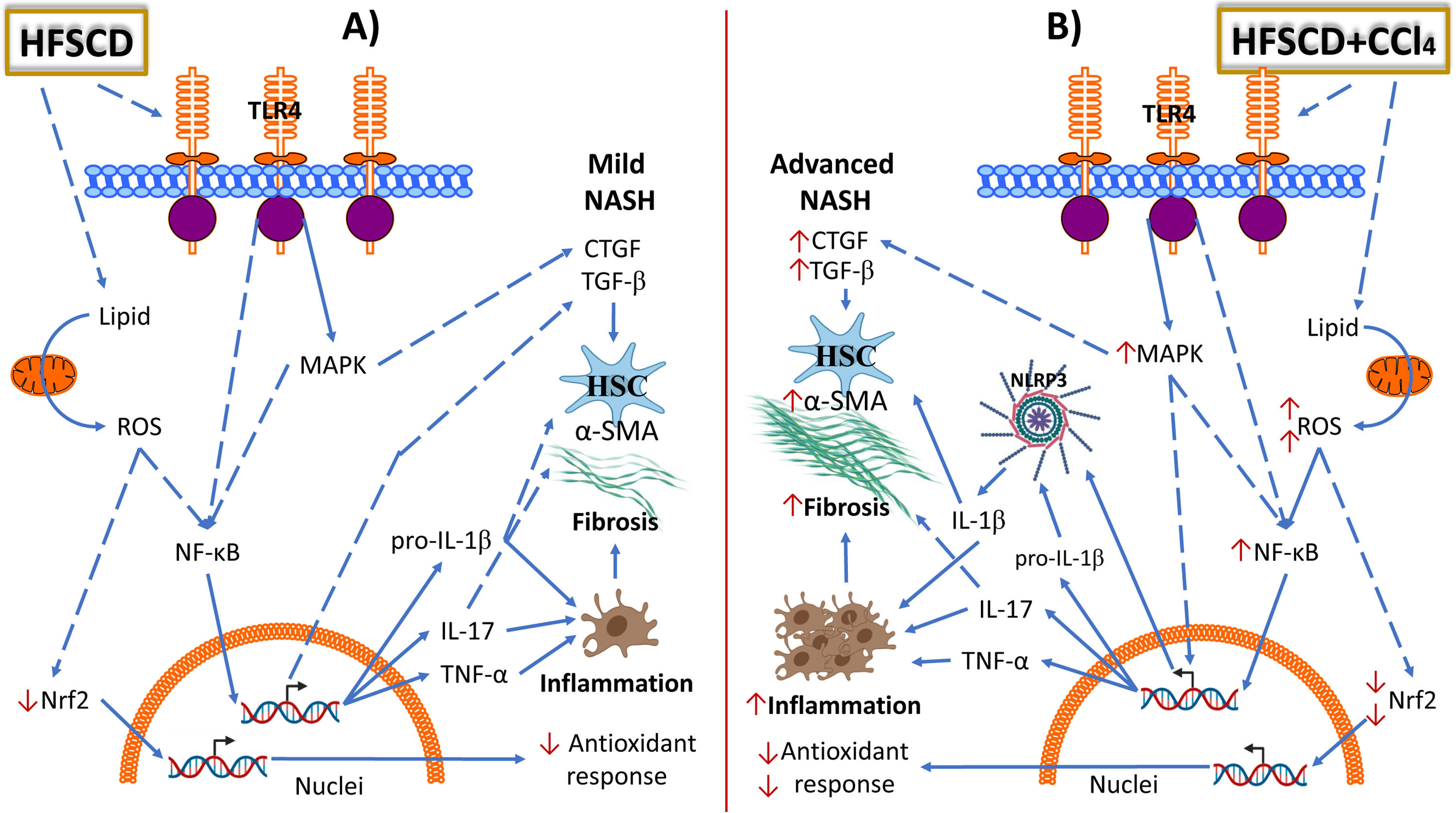
 administration further promotes ROS production in HFSCD-fed rats. HFSCD and HFSCD+CCl4 protocols increase the levels of toll-like receptor 4 (TLR4) and mitogen-activated protein kinase (MAPK) proteins, which trigger the activation of nuclear factor kappa-B (NF-κB), which is the master regulator of inflammation. NF-κB positively regulates NLRP3, IL-1β, IL-17, and TNF-α. Administration of low CCl4 doses to HFSCD-fed rats increases the levels of MAPK and NF-κB compared with those in rats fed HFSCD alone. HFSCD alone does not induce NLRP3 inflammasome activation; however, the concomitant administration of low CCl4 doses to HFSCD-fed rats triggers the activation of proinflammatory pathways. HFSCD and HFSCD+CCl4 protocols activate hepatic stellate cells (HSCs) by inducing transforming growth factor-beta (TGF-β) and connective tissue growth factor (CTGF) signaling. Thus, HFSCD induces inflammation and fibrosis through NF-κB signaling pathway activation; however, when low CCl4 doses are co-administered, the NRLP3 inflammasome pathway is activated to further potentiate inflammation and fibrosis. Solid lines depict direct links and dashed lines indirect ones (involving multiple components, for example, indicates phosphorylation events, activation, etc.). HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, ROS: reactive oxygen species, Nrf2: nuclear factor erythroid 2-related factor 2, NLRP3: nucleotide-binding domain leucine-rich-containing family, IL: interleukin, TNF-α: tumor necrosis factor-alpha, MAPK: mitogen-activated protein kinase, α-SMA: smooth muscle alpha-actin.")
Schematic representation of the effects of HFSCD and HFSCD+CCl4 protocols in the rat liver. Panel A shows the molecular events induced by 15 weeks of treatment with the hepatopathogenic diet. Panel B shows the molecular events induced by 15 weeks of treatment with the hepatopathogenic diet plus weekly low doses of CCl4. HFSCD and HFSCD+CCl4 protocols lead to the accumulation of lipids in liver tissue, exacerbate fatty acid oxidation, increase ROS production, and decrease Nrf2 protein levels. Carbon tetrachloride (CCl4) administration further promotes ROS production in HFSCD-fed rats. HFSCD and HFSCD+CCl4 protocols increase the levels of toll-like receptor 4 (TLR4) and mitogen-activated protein kinase (MAPK) proteins, which trigger the activation of nuclear factor kappa-B (NF-κB), which is the master regulator of inflammation. NF-κB positively regulates NLRP3, IL-1β, IL-17, and TNF-α. Administration of low CCl4 doses to HFSCD-fed rats increases the levels of MAPK and NF-κB compared with those in rats fed HFSCD alone. HFSCD alone does not induce NLRP3 inflammasome activation; however, the concomitant administration of low CCl4 doses to HFSCD-fed rats triggers the activation of proinflammatory pathways. HFSCD and HFSCD+CCl4 protocols activate hepatic stellate cells (HSCs) by inducing transforming growth factor-beta (TGF-β) and connective tissue growth factor (CTGF) signaling. Thus, HFSCD induces inflammation and fibrosis through NF-κB signaling pathway activation; however, when low CCl4 doses are co-administered, the NRLP3 inflammasome pathway is activated to further potentiate inflammation and fibrosis. Solid lines depict direct links and dashed lines indirect ones (involving multiple components, for example, indicates phosphorylation events, activation, etc.). HFSCD: high-fat, sucrose, and cholesterol diet, CCl4: carbon tetrachloride, ROS: reactive oxygen species, Nrf2: nuclear factor erythroid 2-related factor 2, NLRP3: nucleotide-binding domain leucine-rich-containing family, IL: interleukin, TNF-α: tumor necrosis factor-alpha, MAPK: mitogen-activated protein kinase, α-SMA: smooth muscle alpha-actin.
It would be advisable for future research to sacrifice the experimental animals at different times to identify the different stages of the natural progression of NASH such as steatosis, inflammation, and fibrosis, in addition to investigating what would happen if CCl4 is administered after several weeks of consuming an hepatopathogenic diet, and to evaluate if weeks after suspending the diet or the diet plus CCl4, the experimental animals can recover from the injuries. However, we found that the diet concomitantly administered with low weekly doses of CCl4 for 15 weeks mimics several aspects of human NASH.
5ConclusionWe show that HFSCD concomitantly administered with low doses of CCl4 weekly recapitulates several aspects associated with the pathogenesis of NASH as it occurs in humans, including the activation of the most significant proinflammatory pathways, NF-κB and the NLRP3 inflammasome (Table 1). Additionally, our findings highlight the relevance of activation of the NLRP3 inflammasome in NASH as a molecular mechanism that potentiates the progression to fibrosis. We also developed a NASH model induced by an hepatopathogenic diet plus low CCl4 doses that closely mimics human NASH as a useful tool to recapitulate steatosis, inflammation, and fibrosis, as well as to identify biomarkers and molecular targets and to evaluate therapeutic agents during more severe stages of NASH.
Effects of HFSCD and HFSCD+CCl4 protocols in the rat liver.
ROS: Reactive oxygen species; Nrf2: Nuclear factor erythroid 2-related factor 2; TLR4: toll-like receptor 4; MAPK: Mitogen-activated protein kinase; NF-κB: nuclear factor kappa-B; NLRP3: Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; IL: Interleukin; TNF-α: Tumor necrosis factor-alpha; α-SMA: Smooth muscle alpha-actin; TGF-β: Transforming growth factor-beta; CTGF: Connective tissue growth factor; NASH: nonalcoholic steatohepatitis; ↑: Slight increase; ↑↑↑: Severe increase; ↓: Slight decrease; ↓↓↓: Severe decrease.
Vargas-Pozada EE, Ramos-Tovar E, Rodriguez-Callejas JD, Cardoso-Lezama I, Arellanes-Robledo J, Villa-Treviño S, Vásquez-Garzón VR, performed the biochemical, histological, molecular and zymography determinations; Galindo-Gómez S, Gil-Becerril K, Tsutsumi V, performed the histological staining, electron microscopy and their interpretation; and Muriel P and Vargas-Pozada EE designed the research and wrote the original version of the manuscript.
Financial supportThis research was supported by the National Council of Science and Technology (Conacyt) of Mexico (grant no. 53358 to P. Muriel and A1-S-27705 to V. Tsutsumi, and fellowship no. 724340 to E.E. Vargas-Pozada). Erika Ramos-Tovar and Juan D. Rodriguez-Callejas thank the postdoctoral scholarship from Conacyt.
The authors thank Rosa E. Flores-Beltrán, Laura Dayana Buendia-Montaño, Rafael Leyva, Benjamín E. Chavez, Ricardo Gaxiola and Daniel Talamás Lara for excellent technical assistance. The authors also acknowledge the Animal Lab Facility to UPEAL-Cinvestav and Dr. Jorge Fernández-Hernández.



