



Alternative splicing produces complex and dynamic changes in the protein isoforms that are necessary for the proper biological functioning of the metabolic pathways involved in liver development and hepatocyte homeostasis. Changes in the physiological state of alternatively spliced forms are increasingly linked to liver pathologies. This may occur when the expression or function of the set of proteins controlling the alternative splicing processes are altered by external effectors such as oxidative stress and other environmental variations. Studies addressing these modifications reveal a complex interplay between the expression levels of different proteins that regulate the alternative splicing process as well as the changes in alternative splicing. This interplay results in a cascade of different protein isoforms that correlate with the progression of non-alcoholic fatty liver disease, hepatocellular carcinoma, and alcoholic liver disease. However, research on the detailed molecular mechanism underlying the production of these isoforms is needed. It is imperative to identify the physiological processes affected by the differentially spliced isoforms and confirm their role on the onset and maintenance of the pathology. This is required to design potential therapeutic approaches targeting the key splicing changes to revert the pathological condition as well as identify prognostic markers. In this review, we describe the complexity of the splicing process through an example to encourage researchers to go down this path. Subsequently, rather than a catalog of splicing events we have hand-picked and discuss a few selected studies of specific liver pathologies and suggested ways to focus research on these areas.
The liver is the main metabolic organ in the body and it performs its functions by involving highly complex biochemical pathways. This complexity is found in the set of structural proteins, in the enzymes that drive different reactions, in the multiple receptors for interactions with the body fluids and the maintenance of their homeostasis, storage of lipids and carbohydrates and many other biochemical systems. These create a complex proteome that is derived from a complex transcriptome. Additionally, the liver transcriptome is also flexible and capable of responding to developmental and physiological variations in the environment. Dysregulation of this fine-tuned machinery can result in several pathological processes.
Identifying the basis of transcriptomic variations and their impact on pathogenic processes is difficult. Alterations can be derived from a straightforward mutation affecting mRNA splicing or at the other end of the spectrum, through a combination of subtle differences that either per se or along with the genetic background, alter the functionality of the cell. Individual differences by themselves may not result in pathological changes; however, a combination of several slight deficiencies can result in a serious dysfunction. In fact, a cascade of subtle differences or defects in the splicing process can lead to changes in the balance of isoforms produced from alternative splicing with different functionalities and leading to a less efficient proteome. For example, slightly lower or higher levels of specific splicing factors with a synergistic effects on the inclusion or exclusion of an exon can change the properties of the protein (i.e., membrane bound or not, loss of ligand for a receptor, etc.)
In the life cycle of a cell, RNA processing is in-between DNA transcription and RNA translation into protein. RNA processing, due to the lack of comprehension of its complexity was regarded as the Cinderella of gene expression. Subsequently, 20 years ago, the complete human genome sequence demonstrated that RNA alternative splicing is the fundamental process that provides a highly complex transcriptome from a limited number of genes bringing this process and associated research to the limelight [1].
During RNA splicing, the primary RNA transcript (pre-mRNA), whose nucleotide sequence is copied from DNA by RNA polymerase II (Pol II) in the cell nucleus, undergoes several modifications before being transported into the cytoplasm, where translation into protein takes place. The main modification is to provide continuity to the nucleotides coding for the protein interrupted by introns; this essential step is called pre-mRNA splicing and is carried out by a supramolecular machine called the spliceosome [2].
In addition, alternative splicing, which is an further complexity of pre-mRNA splicing, contributes to the generation of protein diversity by expanding the limited number of gene sequences to a much larger proteome based on different combinations of the sequences in the mRNA from one gene. Alternative splicing may be considered a rule rather than an exception, with more than 90% of multi-exon genes undergoing alternative splicing [3,4]. Differences in pre-mRNA processing can result in different outcomes, and may be used by the cells to downregulate an mRNA through nonsense-mediated decay by inclusion of a “poison” exon that shifts the transcript out of frame or by inclusion of an intronic sequence [5]. Additionally, this may create proteins with different functions [6] or alter their localization and stability [7,8]. A proteome-wide mass spectrometry analysis has estimated that approximately 35–40% of the genes generate multiple protein isoforms [9]. Furthermore, different protein isoforms may be expressed in specific developmental stages or in different tissues, as well as within the same tissue in healthy or pathological conditions.
The number of changes in alternative spliced isoforms associated with liver pathologies identified to date is certainly impressive; however, the specific functional characteristics, if any, of the protein isoforms produced remain unexplored. This encourages further biochemical analyses and investigations to determine the effects of variations leading to different pathologies. Understanding the physiological relevance of alternative splicing events is one of the biggest challenges in molecular biology and molecular pathology. This is particularly pertinent in the liver, where there is a wide functional impact of alternative splicing. In fact, alternative splicing is involved in liver development and maintenance of the differentiated state [10] as well as in governing adaptive changes throughout the life span of the organism, for example, during liver regeneration [11]. Thus, it is not surprising that alternative splicing derangements are associated with liver disease. A better understanding of the splicing mechanism and the networks of tissue splicing are essential for comprehending the basis of many diseases, as their dysfunction has multiple impacts on cell and tissue metabolism.
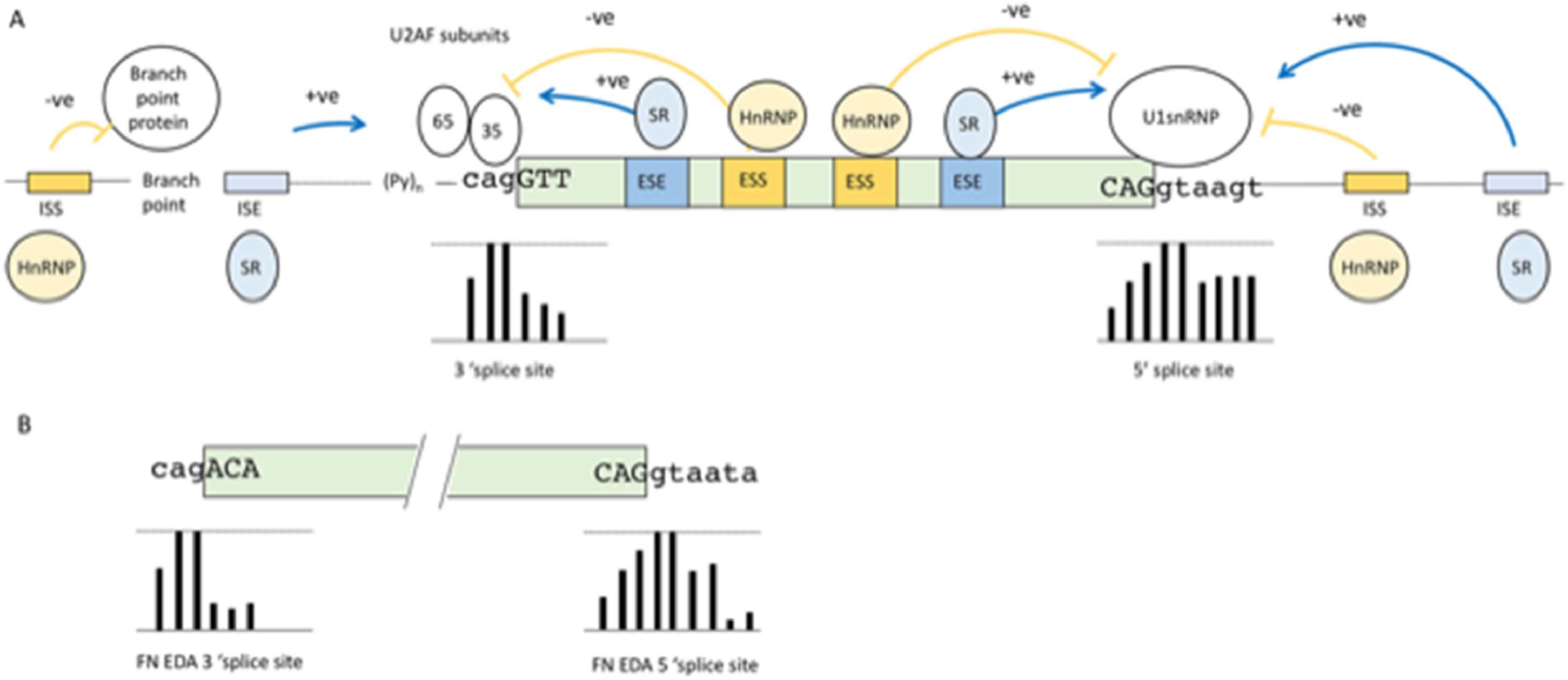
2Basic mechanisms of pre-mRNA splicingBefore discussing diseases that can result from failures in the pre-mRNA splicing process we describe the basic mechanisms of pre-mRNA splicing to understand the points where errors can occur, mutations can be harmful and therapeutic intervention may be attempted. During the pre-mRNA splicing process, introns are removed in a surprisingly precise manner, considering the complexity involved. The catalytic supramolecular complex involved in this process is called the spliceosome. The composition of the spliceosome includes more than 100’s of proteins, many of which are RNA Binding Proteins (RNABP) and RNA-protein complexes called the small nuclear ribonucleoproteins (snRNP). The spliceosome has a constant core, and many additional “players” are present only in specific spliceosome complexes [12]. One of the first steps in the formation of this complex is the recognition of the exon boundaries. This recognition occurs via sequence signals that are highly degenerate [13]. An overall schematic summary of the elements involved is presented in Fig. 1A. The core signals are known as the 5’ and 3’ splice sites. Fig. 1A shows the percentage of frequency of each nucleotide for each position of the splice sites obtained from a collection of over 350,000 splice sites [14]. The consensus sequence of the 5’ splice site is roughly complementary to the 5’ sequence of U1 snRNA, the core component of U1 snRNP. Initial recognition of the 5’ splice site by U1 RNA by base pairing initiates spliceosome formation. However, as seen in Fig. 1A, in the 5’ splice site of an exon, only the first two nucleotides which is the excision site of the intron, downstream of the exon are universally conserved, although a minimum of complementarity is needed and is usually provided by some of the other nucleotides. The second consensus sequence is the loosely defined 3’ splice site region (Fig. 1A). This is composed of three separate elements: the branch site, the polypyrimidine tract, and the 3’splice site. Similar to the 5’ splice site, these also show poor sequence homology at the intron exon junction (Fig 1A).
 Consensus motifs of nucleotide sequences surrounding the intron-exon borders that act as essential splicing signals. Bars below the nucleotides represent the frequency of the nucleotides shown at that position from over 350,000 splices sites, with the top line denoting 100% frequency and bottom line denoting 0% frequency. (B) Splice sites of the EDA exon. Bars below the nucleotides show the frequency of each nucleotide position compared to more than 350,000 splices sites. As can be seen in the 5’ splice site, these deviate significantly from the consensus sequence at the +5 and +6 positions; whereas in the 3’ splice site, these deviate from consensus sequence at the +1 and -3 positions.")
(A) Consensus motifs of nucleotide sequences surrounding the intron-exon borders that act as essential splicing signals. Bars below the nucleotides represent the frequency of the nucleotides shown at that position from over 350,000 splices sites, with the top line denoting 100% frequency and bottom line denoting 0% frequency. (B) Splice sites of the EDA exon. Bars below the nucleotides show the frequency of each nucleotide position compared to more than 350,000 splices sites. As can be seen in the 5’ splice site, these deviate significantly from the consensus sequence at the +5 and +6 positions; whereas in the 3’ splice site, these deviate from consensus sequence at the +1 and -3 positions.
The junction sequence of exons and introns, defined as “weak” splice sites, depart considerably from the consensus sequences. They are found in most genes and are particularly abundant in alternatively spliced exons (see paragraph below) [14]. In these cases, splice site recognition by the spliceosome machinery is aided by auxiliary sequences in the exon itself or in the intron beyond or before the 5’ and 3’ splice site. These auxiliary elements are essential in alternative splicing, and disruption of their binding site by point mutations is often a cause of disease [15]. These auxiliary elements are also degenerate and difficult to identify, and consequently present a great challenge for clinical geneticists [16]. The auxiliary elements can be grouped into four types, although elements with dual modes of action also exist [17]: exonic splicing enhancers (ESE), intronic splicing enhancers (ISE), exon splicing silencers (ESS) and intronic splicing silencers (ISS) see Fig. 1A. These cis-acting elements serve as binding sites for a large family of structurally related RNABPs. Generally, ESE and ISE bind to serine/arginine-rich (SR) proteins [18]. These proteins share a common structure consisting of RNA-recognition motifs (RRM) and by a distinctive C-terminal domain that is highly enriched in serine-arginine dipeptides. The RRMs bind to the RNA through degenerate target sequences, whereas the serine-arginine rich domain is involved in protein-protein interactions and promotes exon definition by directly recruiting the spliceosome machinery [19]. The negative elements balance the positive effect of enhancers; the ESS and ISS are usually the binding sites of heterogeneous nuclear ribonucleoproteins (hnRNPs), which are a set of primarily nuclear proteins that bind pre-mRNA through one or more RRMs without forming stable associations with other RNA-protein complexes [20]. The mechanisms by which ESS and ISS interfere with splicing include blocking the access of the spliceosome to the splice sites by steric hindrance and/or competing with positive factors (SR proteins) for the same binding site. According to the levels of the positive and negative splicing factors in these physiological conditions, the final decision about exon inclusion is made by a balance of power between the specific binding affinities and abundance of the positive and negative factors [21]. The enhancer and silencer sequences increase or decrease the specificity of the exon boundary definition [22]. The degeneracy of these sequences as well as the splice sites is difficult to understand for such a precise mechanisms. However, it allows RNA splicing to be flexible and capable of performing alternative splicing.
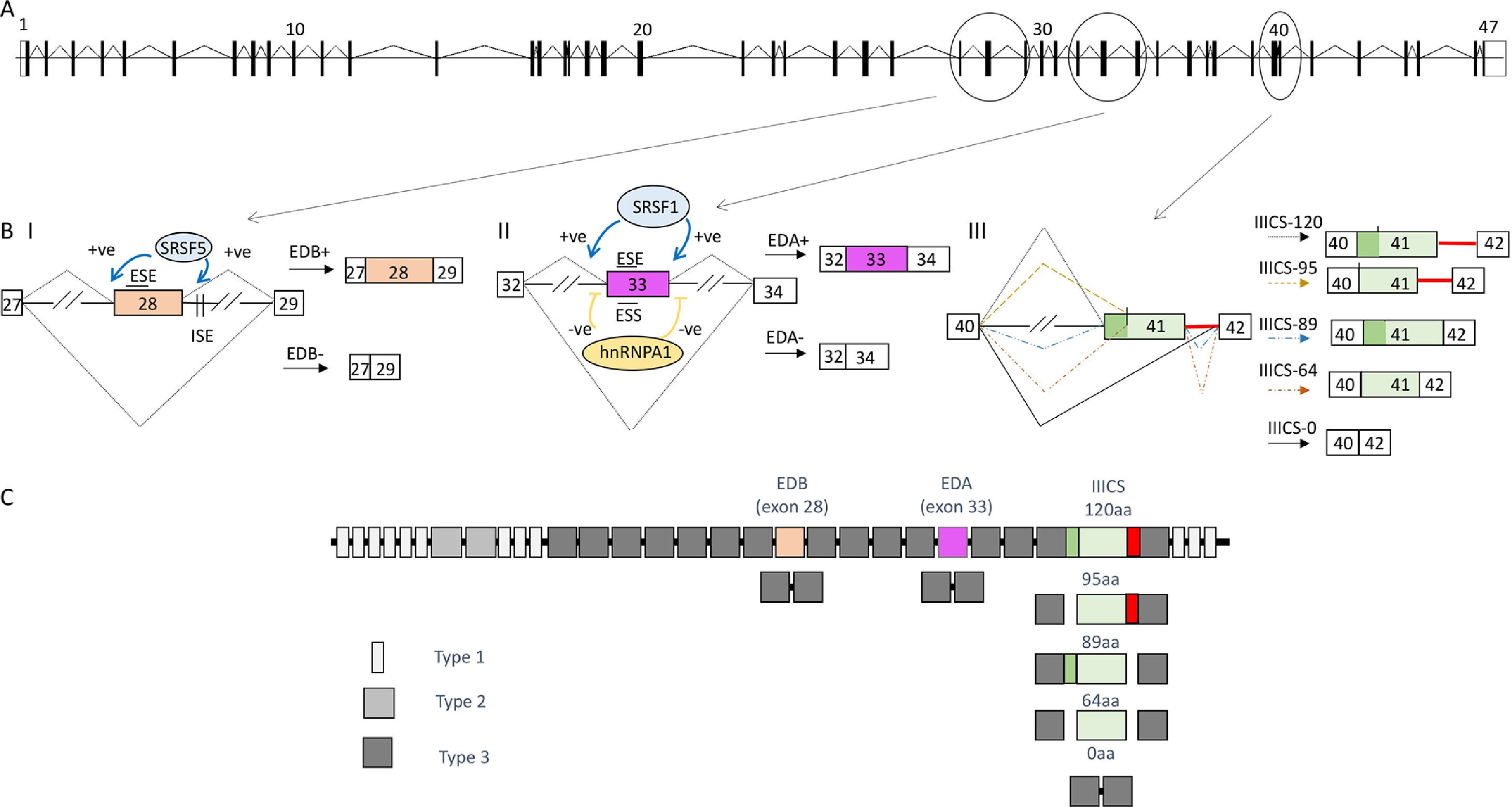
As above mentioned, alternative splicing is a process in which a range of different mRNAs and proteins can be produced from a single gene. There are several different types of alternative splicing; the most common form in the human genome is cassette exon alternative splicing where an intervening exon (between two other exons) from the mature mRNA sequence is partially included or skipped to generate two distinct protein isoforms. Other modes include mutually exclusive alternative splicing, where only one of two or more exons is included in the mRNA; the use of alternative 5’ and 3’ splice sites that result in intron retention (either completely or part thereof); and shortening or lengthening of an exon. Furthermore, different promoters and different polyadenylation sites may specify respectively, alternative 5’ and 3’ terminal exons, respectively. An example of medium complexity, the Fibronectin (FN) gene system and the mechanisms involved is shown in Fig. 2. The legend explains in detail the events and mechanisms observed in this gene.
![(A) Diagram depicting, approximate in scale the FN gene based on the ensemble reference sequence transcript ID ENST00000323926.10. The boxes represent the 47 exons, separated by intervening sequences (introns), which are shown as lines. Constitutive splicing is indicated by sloping lines (B) Alternative splicing of the exons EDB, EDA and IIICS region. Splicing is indicated by sloping lines. Trans-acting factors that bind to the enhancer and silencer elements in the exons (ESE; ESS) and/or introns (ISE; ISS) as well as their effect (-ve = inhibitory, +ve = enhancement) are indicated. // indicates that the intronic length is longer and a section has been eliminated to fit the figure. (BI) FN-EDB cassette exon inclusion is dependent on intronic splicing enhancers (ISE) downstream of the EDB [74] that are recognized by the splicing factor SRSF5. It has further been observed that SRSF5 levels are upregulated during liver cell proliferation, during development, and liver regeneration [75]. The factor SRSF5 promotes EDB inclusion in regenerating liver by binding an exonic splicing enhancer within the exon per se [76]. This exon is included mostly during development or tumor transformation. (BII) The FN-EDA 5’ss splice sites and 3’ splice sites deviates considerably from the consensus sequence (Fig. 1 B shows comparison with the consensus sequence in Fig. 1 A). The fact that the EDA exon has these weak (non-consensus) splice sites means that it is also strongly subjected to the effect of the auxiliary acting element. In fact EDA exon inclusion is stimulated by the binding of the splicing factor SRSF1 [77] to an ESE element characterized in detail within exon 33 of the pre-mRNA that encodes the EDA [78]. In the case of EDA exon skipping this is, in part, because of binding sites within the exon for hnRNP A1 [79]. (BIII) Alternative splicing of the IIICS region of the FN gene. This region is less well characterized in terms of the molecular mechanisms dictating the choice of the alternative exons. These are more complex alternatively spliced forms and a perfect example of the different types of alternative splicing mentioned above. Alternative splicing is centered in the sequence spanning exon 41 and the intronic sequence downstream. In humans, there are five different splicing variations of this region [80], and the manner by which these arise are outlined. Briefly, retention of the intron downstream of exon 41 is responsible for the production of the larger isoform corresponding to IIICS-120. The IIICS 95 isoform is produced by an alternative 3’ splice site within exon 41 together with the inclusion of the downstream intron. The IIICS 89 isoform on the other hand may be considered to follow a “normal splicing” of the introns. The IIICS 64 isoform is produced from the internal 3’ splice site in exon 41; however, this time the downstream intron been is not included. Finally, the IIICS 0 isoform is produced by skipping of exon 41. This type of complex splicing pattern likely uses all possible auxiliary elements enhancers, ESE and ISE, and silencers, ESS and ISS, to enhance and repress the alternative 5’ and 3’ splices sites used. The silencers elements repress the splice site choice through several different mechanisms. For example, they may block spliceosome activity via steric hindrance by blocking the access to the splice sites and/or competing with positive factors (SR proteins) for the same binding site. According to the levels of the positive and negative splicing factors in those physiological conditions the final decision about exon inclusion is made by a balance of power between the specific binding affinities of the positive and negative factors [21]. In the case of the IIICS alternative splicing, for example it is known to date that a splicing factor known as SWAP promotes the exclusion of the entire IIICS region [81]. Although the location where this protein binds is yet to be determined. In the same study SRSF1 was also identified to promote the full inclusion of the IIICS region; however the cis-acting elements are yet to be identified. (C) The different types of homologies (12 Type I, 2 Type II and 15 Type III) are represented.](https://static.elsevier.es/multimedia/16652681/000000260000000C/v5_202208200714/S1665268121002337/v5_202208200714/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9DlLsuks/7UI2O056dpyiniMrPtzOVwfdjL2SljRncH9QQtUnuq8Spt0pThzW9ZdlKpbCL3hnc8b0ScnXTR9e5dWeicuNMw7OdL1WaHbWK/EhvB7mjEiyl44hVeWbLG6yFtO1VBZlJRlJZG5EBqyeF+QZbV2a2MbxKUxsa4Shvg4Wla7aCyEI+VZefqeAyIYJeM2KlE91M8bc1WODP33o65QsTdjj5w+68BZQUO86rfxX4= "(A) Diagram depicting, approximate in scale the FN gene based on the ensemble reference sequence transcript ID ENST00000323926.10. The boxes represent the 47 exons, separated by intervening sequences (introns), which are shown as lines. Constitutive splicing is indicated by sloping lines (B) Alternative splicing of the exons EDB, EDA and IIICS region. Splicing is indicated by sloping lines. Trans-acting factors that bind to the enhancer and silencer elements in the exons (ESE; ESS) and/or introns (ISE; ISS) as well as their effect (-ve = inhibitory, +ve = enhancement) are indicated. // indicates that the intronic length is longer and a section has been eliminated to fit the figure. (BI) FN-EDB cassette exon inclusion is dependent on intronic splicing enhancers (ISE) downstream of the EDB [74] that are recognized by the splicing factor SRSF5. It has further been observed that SRSF5 levels are upregulated during liver cell proliferation, during development, and liver regeneration [75]. The factor SRSF5 promotes EDB inclusion in regenerating liver by binding an exonic splicing enhancer within the exon per se [76]. This exon is included mostly during development or tumor transformation. (BII) The FN-EDA 5’ss splice sites and 3’ splice sites deviates considerably from the consensus sequence (Fig. 1 B shows comparison with the consensus sequence in Fig. 1 A). The fact that the EDA exon has these weak (non-consensus) splice sites means that it is also strongly subjected to the effect of the auxiliary acting element. In fact EDA exon inclusion is stimulated by the binding of the splicing factor SRSF1 [77] to an ESE element characterized in detail within exon 33 of the pre-mRNA that encodes the EDA [78]. In the case of EDA exon skipping this is, in part, because of binding sites within the exon for hnRNP A1 [79]. (BIII) Alternative splicing of the IIICS region of the FN gene. This region is less well characterized in terms of the molecular mechanisms dictating the choice of the alternative exons. These are more complex alternatively spliced forms and a perfect example of the different types of alternative splicing mentioned above. Alternative splicing is centered in the sequence spanning exon 41 and the intronic sequence downstream. In humans, there are five different splicing variations of this region [80], and the manner by which these arise are outlined. Briefly, retention of the intron downstream of exon 41 is responsible for the production of the larger isoform corresponding to IIICS-120. The IIICS 95 isoform is produced by an alternative 3’ splice site within exon 41 together with the inclusion of the downstream intron. The IIICS 89 isoform on the other hand may be considered to follow a “normal splicing” of the introns. The IIICS 64 isoform is produced from the internal 3’ splice site in exon 41; however, this time the downstream intron been is not included. Finally, the IIICS 0 isoform is produced by skipping of exon 41. This type of complex splicing pattern likely uses all possible auxiliary elements enhancers, ESE and ISE, and silencers, ESS and ISS, to enhance and repress the alternative 5’ and 3’ splices sites used. The silencers elements repress the splice site choice through several different mechanisms. For example, they may block spliceosome activity via steric hindrance by blocking the access to the splice sites and/or competing with positive factors (SR proteins) for the same binding site. According to the levels of the positive and negative splicing factors in those physiological conditions the final decision about exon inclusion is made by a balance of power between the specific binding affinities of the positive and negative factors [21]. In the case of the IIICS alternative splicing, for example it is known to date that a splicing factor known as SWAP promotes the exclusion of the entire IIICS region [81]. Although the location where this protein binds is yet to be determined. In the same study SRSF1 was also identified to promote the full inclusion of the IIICS region; however the cis-acting elements are yet to be identified. (C) The different types of homologies (12 Type I, 2 Type II and 15 Type III) are represented.")
(A) Diagram depicting, approximate in scale the FN gene based on the ensemble reference sequence transcript ID ENST00000323926.10. The boxes represent the 47 exons, separated by intervening sequences (introns), which are shown as lines. Constitutive splicing is indicated by sloping lines (B) Alternative splicing of the exons EDB, EDA and IIICS region. Splicing is indicated by sloping lines. Trans-acting factors that bind to the enhancer and silencer elements in the exons (ESE; ESS) and/or introns (ISE; ISS) as well as their effect (-ve = inhibitory, +ve = enhancement) are indicated. // indicates that the intronic length is longer and a section has been eliminated to fit the figure. (BI) FN-EDB cassette exon inclusion is dependent on intronic splicing enhancers (ISE) downstream of the EDB [74] that are recognized by the splicing factor SRSF5. It has further been observed that SRSF5 levels are upregulated during liver cell proliferation, during development, and liver regeneration [75]. The factor SRSF5 promotes EDB inclusion in regenerating liver by binding an exonic splicing enhancer within the exon per se [76]. This exon is included mostly during development or tumor transformation. (BII) The FN-EDA 5’ss splice sites and 3’ splice sites deviates considerably from the consensus sequence (Fig. 1 B shows comparison with the consensus sequence in Fig. 1 A). The fact that the EDA exon has these weak (non-consensus) splice sites means that it is also strongly subjected to the effect of the auxiliary acting element. In fact EDA exon inclusion is stimulated by the binding of the splicing factor SRSF1 [77] to an ESE element characterized in detail within exon 33 of the pre-mRNA that encodes the EDA [78]. In the case of EDA exon skipping this is, in part, because of binding sites within the exon for hnRNP A1 [79]. (BIII) Alternative splicing of the IIICS region of the FN gene. This region is less well characterized in terms of the molecular mechanisms dictating the choice of the alternative exons. These are more complex alternatively spliced forms and a perfect example of the different types of alternative splicing mentioned above. Alternative splicing is centered in the sequence spanning exon 41 and the intronic sequence downstream. In humans, there are five different splicing variations of this region [80], and the manner by which these arise are outlined. Briefly, retention of the intron downstream of exon 41 is responsible for the production of the larger isoform corresponding to IIICS-120. The IIICS 95 isoform is produced by an alternative 3’ splice site within exon 41 together with the inclusion of the downstream intron. The IIICS 89 isoform on the other hand may be considered to follow a “normal splicing” of the introns. The IIICS 64 isoform is produced from the internal 3’ splice site in exon 41; however, this time the downstream intron been is not included. Finally, the IIICS 0 isoform is produced by skipping of exon 41. This type of complex splicing pattern likely uses all possible auxiliary elements enhancers, ESE and ISE, and silencers, ESS and ISS, to enhance and repress the alternative 5’ and 3’ splices sites used. The silencers elements repress the splice site choice through several different mechanisms. For example, they may block spliceosome activity via steric hindrance by blocking the access to the splice sites and/or competing with positive factors (SR proteins) for the same binding site. According to the levels of the positive and negative splicing factors in those physiological conditions the final decision about exon inclusion is made by a balance of power between the specific binding affinities of the positive and negative factors [21]. In the case of the IIICS alternative splicing, for example it is known to date that a splicing factor known as SWAP promotes the exclusion of the entire IIICS region [81]. Although the location where this protein binds is yet to be determined. In the same study SRSF1 was also identified to promote the full inclusion of the IIICS region; however the cis-acting elements are yet to be identified. (C) The different types of homologies (12 Type I, 2 Type II and 15 Type III) are represented.
In the following sections, we will describe the analysis and biological effects of alternative isoforms of the FN gene, as well as introduce alternative splicing and its mechanisms. Previous reviews provide a more comprehensive understanding of alternative splicing [23–26]. After this example, we will describe other splicing events and/or splicing networks involved in liver pathologies such as Nonalcoholic Fatty Liver Disease (NAFLD), Hepatocellular carcinoma (HCC) and Alcoholic Liver Disease (ALD).
3Fibronectin and the biological role of its isoformsFNs are ubiquitous multifunctional, and high molecular weight proteins that play various roles in cellular interactions with the extracellular matrix as well as in cell migration, growth and differentiation [27]. FN undergoes alternative splicing, generating up to 20 different variants in humans. This complex splicing pattern is particularly centered in three regions of its pre-mRNA, Fig. 2 describes these in detail to explain alternative splicing in a more comprehensive manner to the reader and in particular that of alternative splicing of the cassette EDA exon (extra type III domain). This FN alternative splicing was one of the first cases to be described [28]; the regulatory mechanisms of the same have been studied in depth since the mid-eighties making it a good example of alternative splicing of a cassette exon.
The descriptive collection of alternative splicing isoforms and the mechanism by which they are produced is the first step in analyzing the biological functions. Uncovering the latter is a laborious process and often identifying the biological roles for alternative spliced isoforms deriving from transcriptome analysis are put aside. Understanding the exact biochemical role of alternatively spliced isoforms is essential in order to better understand their biological significance and their place in any eventual pathological condition. The following example illustrates the studies performed to establish the biological role of the FN isoforms.
Circulating soluble plasma FN secreted by hepatocytes generally lacks both the EDA and EDB segments, whereas cellular FN contains variable proportions of EDA, EDB, or both, and are enriched in the extracellular matrix [29]. FN-EDA and FN-EDB are nearly ubiquitously expressed in embryonic tissues. Specific IIICS isoforms are found exclusively in the cartilage [29], and 50% of FN in the plasma may lack the IIICS domain These expression patterns vary in many physiological or pathological states; for example, FN-EDB is upregulated during embryogenesis and is temporally activated during wound healing, tissue repair, and angiogenesis [30]. FN-EDB levels are elevated in the cerebrospinal fluid of patients with bacterial meningitis. The presence of the EDB domain in FN enhances phagocytic response towards bacteria via αvβ3 integrin [31]. In contrast, certain FN-IIICs isoforms are altered after peripheral nerve injury [32]. In the case of FN, spliced forms of the EDA exon are the better understood, mostly because of the establishment of a transgenic mice that are unable to perform specific alternative splicing of the EDA [33]. In this study, the physiological gene regulation was kept intact as mice constitutive EDA exon inclusion was obtained by optimizing the splice sites by using Cre-loxP knock in technology, whereas complete exclusion was obtained after in vivo Cre-loxP–mediated deletion of the exon.
Although homozygous mice with inclusion or exclusion of the EDA exon develop normally, both mouse strains which lack splicing regulation, have a shorter lifespan than the wild-type mice. Further investigations have identified wound healing and fibroblast migration to be suboptimal in mice lacking the EDA exon. This effect may be partly due to the fact that the FN-EDA exon contains binding sites for integrins [34], a family of receptors that control cell adhesion by binding to extracellular matrix ligands, cell-surface ligands, and soluble ligands [35]. In contrast, mice that have FN with constant EDA inclusion showed a considerable decrease in the FN levels. This data indicate that EDA inclusion or skipping modulates FN function. Other roles of the EDA exon have also been uncovered through use of these animals. Mice constitutively expressing the EDA domain are more prone to developing bone marrow fibrosis upon treatment with the thrombopoietin (TPO) mimetic drug romiplostim than those that do not express the EDA exon [36]. FN-EDA stimulates megakaryocytes, which in turn increase the synthesis of proinflammatory cytokines, highlighting a potential therapeutic approach for patients with high-grade myelofibrosis. Another observation as well as an example of the need for in-depth investigations of the isoforms function is from a study into the role of FN-EDA in the development of atherosclerosis [37]. The researchers crossed Apo E-deficient mice, a standard model of atherosclerosis with mice lacking the EDA exon to obtain the Apo ED−/−, EDA−/− genotype. It was originally suggested that FN-EDA has a pro-atherogenic role, as these mice showed reduced atherosclerosis after an atherogenic-diet [38]. A subsequent study showed that this was occurring via Toll like receptor 4 signaling, a receptor involved in innate immunity [39], activation through the EDA domain [40], that was in turn enhancing recruitment of monocytes/macrophages into developing plaques [41]. However, constitutive absence or inclusion of the FN-EDA exon was observed to result in decreased levels of atherosclerosis. This raises the intriguing possibility that regulation of the EDA domain by alternative splicing may have role in the progression of the atherogenic process [37].
Finally, FN-EDA has been shown to have an important role in the fibrotic process in many tissues. In particular, the transgenic mice lines, EDA-/- were treated with bleomycin to induce lung fibrosis. This experiment showed that EDA-/- mice failed to develop significant fibrosis while their wild-type littermates did. Failure to develop lung fibrosis in EDA null mice correlated with diminished activation of latent transforming growth factor TGF-b and decreased lung fibroblast responsiveness to active TGF-b in vitro. Unfortunately, the precise role of FN-EDA in hepatic fibrosis still needs to be elucidated. Indeed while FN-EDA has been shown to be upregulated by TGF-β in a hepatic fibrosis model [42] and FN to be essential per se [43], other studies indicate that pathways independent of FN can also result in liver fibrosis [44]. Interestingly FN-EDA has also been observed to play a role in angiogenesis in hepatic fibrosis, by promoting the phosphorylation of vascular endothelial growth factor 2 and could be a potential therapeutic target for the disease [45].
4Fibronectin as a predictor of fibrosis development in NAFLD and chronic Hepatitis C infectionThe role of specific FN isoforms in fibrogenesis (as mentioned above in the case of lung fibrosis) suggests that they may have a similar role in liver fibrosis. This would, in turn, increase ways to use FN measurements as predictor of liver fibrosis. However, definitive evidence to ascribe a pathological role in liver fibrosis to specific FN isoforms is still in need of further investigation; this is notwithstanding the attempts that have used FN levels as a marker for evaluating the development of liver fibrosis. For example, difference in total FN levels between the first and second liver biopsy in a set of patients with non-alcoholic steatohepatitis (NASH), who were followed up for approximately 7 years, was observed to be a good predictor of fibrosis progression (odds 14:1) [46]. In fact, most patients with a certain degree of fibrosis development (approximately half) show an increased level of total FN.
Another study focused on chronic hepatitis C infections used a blood test based on the plasma levels of FN, aspartate aminotransferase to platelet ratio index, and albumin to establish the FN Discriminating Score [47]. This index can predict the risk of having significant liver fibrosis with a better accuracy than the aspartate aminotransferase to to platelet ratio index alone, thereby diminishing the need for repeated liver biopsies.
The factors influencing the development of a disease are difficult to identify, as each factor often contributes only partially to the defect and in isolation cannot produce a visible phenotype. Alternative splicing can result in a slightly different balance of isoforms that are damaging in an age-dependent manner and can contribute to a disease in an almost invisible manner. An example of this can be seen in the FN transgenic animals described above, wherein they show a drastic change in alternative splicing regulation as they do not have the capacity to modulate the inclusion or exclusion of the exon EDA; therefore they are either 100% EDA+ or 100%EDA-. Such animals develop and reproduce with no apparent problem, however a gross phenotype, such as the life span, is reduced in both the transgenic strains. This means that the EDA isoform balance modulates processes that affect longevity; however, not all of them have yet been identified.
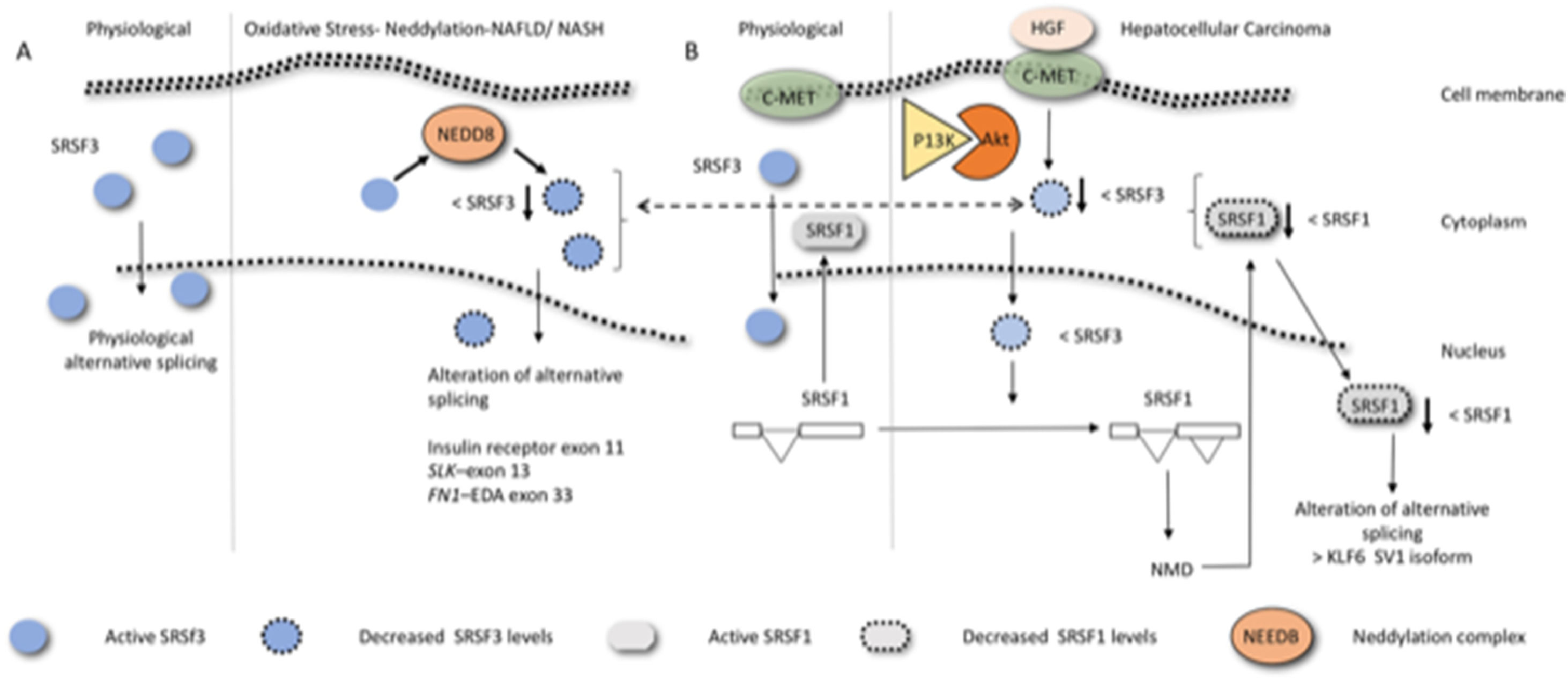
5Non-alcoholic fatty liver disease and alternative splicingThe FN example shows clearly why the regulation of alternative splicing has emerged in recent years as a promising research target in many fields including obesity, with current studies focusing on identifying the obesity-related alternative splicing events [48]. Obesity is a multifactorial disease influenced both by the genetic makeup of an individual and the environment. NAFLD/MAFLD, which is strongly associated with obesity, is a series of conditions that lead to the build-up of excessive fat in the liver, excluding those induced by alcohol extending. The condition ranges from benign simple fatty liver, also known as nonalcoholic fatty liver (NAFL), with no inflammation of the liver, to more serious conditions including liver inflammation and fibrosis, such as NASH and HCC [49]. Interestingly, only a proportion of patients with NAFLD show progress to NASH, fibrosis, and eventually cirrhosis. Although the molecular mechanisms underlying the heterogeneity in the outcome or transformation of NAFLD to more severe pathology remains unsolved, alternative splicing has recently been implicated in this process. Liver disease is generally associated with altered gene expression [50] and altered gene splicing [51]. In the livers of patients with obesity with steatosis, patient-dependent alterations of the cellular machinery responsible for the regulation of both core splicing components and auxiliary splicing factors have been observed [52]. This is interesting because the genetic heterogeneity in the human population due to differences in protein isoforms levels may, therefore, also contribute to the disease progression and thus represent an eventual form of biomarker for clinical evaluation, prognosis prediction, and/or as an eventual therapeutic target. Many of these changes have been ascribed either directly or indirectly to variations in the splicing factor SRSF3. SRSF3 is generally associated with helping in the inclusion of alternative exons. In hepatic fibrosis, and in liver inflammation, this factor has been observed to undergo a reduction of its levels resulting from its degradation by the proteasome via neddylation (Fig. 3A) caused by lipid-induced oxidative stress [51]. Indeed, prevention of SRSF3 degradation in vivo partially protected mice from hepatic steatosis, fibrosis, and inflammation [51] and hepatocyte-specific deletion of the SRSF3 gene in mice has been observed to result in HCC [53]. Exons from different genes whose inclusion or exclusion are known to be dependent on SRSF3 are also altered in human NAFLD, NASH, and liver cirrhosis. For example, exclusion or inclusions of the exon 11 of the insulin receptor (IR) gives rise to the receptor isoforms; IR-A, which excludes exon 11 and IR-B, which includes it. IRA has been reported to be the predominant IR isoform expressed in cancer cells, such as in a variety of carcinomas or breast cancer cell lines [54].This exon inclusion is dependent on SRSF3 [55]. The exon 11- isoform (IRA) binds both insulin and IGF-II with higher affinity that the 11+ (IRB) [56]. Interestingly, the isoform produced to a greater extent upon reduction of SRSF3, IRA, increases glucose uptake, ameliorates insulin sensitivity, and reduces hepatic lipid accumulation, thereby alleviating one of the main pathologies associated with obesity [57]. This is contrary to the pathology expected in a NAFLD and NASH liver. SRSF3 reduction may simultaneously, therefore, result in different isoform, some of which may be protective and some may be harmful to the liver. Such contrasting behavior emphasize the need for biochemically analyzing the function of alternative spliced isoforms to facilitate therapeutic research.
![Diagram exemplifying how alteration of SRSF3 can result in liver pathologies. (A) hepatic fibrosis/ liver inflammation reduces the expression levels of SRSF3 via neddylation. Decreased SRSF3 expression in the nucleus (blue circle with dotted circumference) affects the inclusion or exclusion of hundreds of exons, a few, of which are known to be central to NAFLD, NASH, and liver cirrhosis liver are indicated. (B) Hepatocyte growth factor, binding to hepatocyte growth factor receptor (C-Met), induces the PI3K/Akt signaling pathway to decrease SRSF3 levels. The decrease in SRSF3 levels results in an alternative splicing event within the SRSF1 gene producing an isoform that undergoes nonsense-mediated decay. This results in a decreased in SRSF1 expression. Low expression of SRSF1 in the nucleus produces a variety of alterations in the splicing events among which that of the tumor suppressor Kruppel-like factor 6 (KLF6), enhancing the splicing pattern SV1 isoform [66], which leads to increased cell proliferation as it antagonizes the ability of wild-type KLF6 to suppress it.](https://static.elsevier.es/multimedia/16652681/000000260000000C/v5_202208200714/S1665268121002337/v5_202208200714/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9DlLsuks/7UI2O056dpyiniMrPtzOVwfdjL2SljRncH9QQtUnuq8Spt0pThzW9ZdlKpbCL3hnc8b0ScnXTR9e5dWeicuNMw7OdL1WaHbWK/EhvB7mjEiyl44hVeWbLG6yFtO1VBZlJRlJZG5EBqyeF+QZbV2a2MbxKUxsa4Shvg4Wla7aCyEI+VZefqeAyIYJeM2KlE91M8bc1WODP33o65QsTdjj5w+68BZQUO86rfxX4= "Diagram exemplifying how alteration of SRSF3 can result in liver pathologies. (A) hepatic fibrosis/ liver inflammation reduces the expression levels of SRSF3 via neddylation. Decreased SRSF3 expression in the nucleus (blue circle with dotted circumference) affects the inclusion or exclusion of hundreds of exons, a few, of which are known to be central to NAFLD, NASH, and liver cirrhosis liver are indicated. (B) Hepatocyte growth factor, binding to hepatocyte growth factor receptor (C-Met), induces the PI3K/Akt signaling pathway to decrease SRSF3 levels. The decrease in SRSF3 levels results in an alternative splicing event within the SRSF1 gene producing an isoform that undergoes nonsense-mediated decay. This results in a decreased in SRSF1 expression. Low expression of SRSF1 in the nucleus produces a variety of alterations in the splicing events among which that of the tumor suppressor Kruppel-like factor 6 (KLF6), enhancing the splicing pattern SV1 isoform [66], which leads to increased cell proliferation as it antagonizes the ability of wild-type KLF6 to suppress it.")
Diagram exemplifying how alteration of SRSF3 can result in liver pathologies. (A) hepatic fibrosis/ liver inflammation reduces the expression levels of SRSF3 via neddylation. Decreased SRSF3 expression in the nucleus (blue circle with dotted circumference) affects the inclusion or exclusion of hundreds of exons, a few, of which are known to be central to NAFLD, NASH, and liver cirrhosis liver are indicated. (B) Hepatocyte growth factor, binding to hepatocyte growth factor receptor (C-Met), induces the PI3K/Akt signaling pathway to decrease SRSF3 levels. The decrease in SRSF3 levels results in an alternative splicing event within the SRSF1 gene producing an isoform that undergoes nonsense-mediated decay. This results in a decreased in SRSF1 expression. Low expression of SRSF1 in the nucleus produces a variety of alterations in the splicing events among which that of the tumor suppressor Kruppel-like factor 6 (KLF6), enhancing the splicing pattern SV1 isoform [66], which leads to increased cell proliferation as it antagonizes the ability of wild-type KLF6 to suppress it.
Splicing alterations are considered a hallmark of cancer [58], and are often linked to mutations in genes encoding core components or regulators of the splicing machinery. Thus understanding and characterizing the alterations is important, as they may be used to predict the course of the disease and be used as a route of therapeutic intervention. HCC, which usually results from alcoholic steatohepatitis, NASH, or hepatitis C viral infection, is not an exception to this rule, as highlighted in a recent review [59]. Indeed, because of the central role that alternative splicing may play in the HCC, transcriptome-wide-analysis have been performed on aberrant alternative landscape events in HCC [60–62] and have revealed some interesting features. The alternatively spliced exons that are observed vary greatly among individuals; emphasizing the difficulty in interpreting the effects of isoforms due to the heterogeneity of the human population, and a myriad of regulators of alternative splicing are differentially expressed in HCC. For example, an HCC-based study identified, 53 splicing factors with altered composition changes and 34,000 different alternative splicing alterations from approximately 9000 genes, [63]. This large number cannot all be ascribed as causative of tumor transformation and proliferation or consequence of tumor transformation and proliferation. In fact, the alternative splicing events were quite heterogenic in terms of the expression levels in different individuals. Focusing on the splicing factors the study, additionally identified, PCBP2, SFPQ, and SRSF10 to be associated with a significant adverse prognostic value, indicating that these factors may be affecting the alternative splicing process producing isoforms with a role in HCC, as well as providing a useful basis for initiating prognostic and eventual molecule-targeted studies. In this regard recent studies stratifying HCC patients according to alternative spliced isoforms, for example, resulted in four clusters with different survival patterns [64]. However, future research must concentrate on the physiological role of the differentially spliced isoforms and their connection with the identified variations in the splicing factors.
A rather elegant study along these lines and further stressing the role of the alternative splicing process in HCC regards the splicing factors SRSF3 and SFRS1 [65]. As shown in Fig. 3, hepatocyte growth factor binding to hepatocyte growth factor receptor (c-Met), induces the PI3K/Akt signaling pathway and has been observed to decrease SRSF3 levels. The decrease in SRSF3 expression in turn downregulates SRSF1 levels by alternative splicing within the SRSF1 gene, resulting in an isoform that undergoes nonsense-mediated decay. Decreased SRSF1 expression then alters the alternative splicing of the tumor suppressor Kruppel-like factor 6, enhancing the production dominant-negative splice form KLF6-SV1 [66], which antagonizes the ability of wild-type KLF6 to suppress cell proliferation. SRSF2, another splicing factor, upregulated in HCC [67]; is associated with poor prognosis and affects the splicing of several exons in different genes. In this study specific exclusion or inclusion of a given exon in some of these alternative splicing events can promote the tumorigenic process, while the counterpart isoform (+ or- according to the specific case) does not.
Elucidating the network of disease-related changes in splicing factors, alternative splicing events, and molecular mechanisms will yield the reliable predictors and therapeutic targets. We have arrived to a moment where we are currently looking at the forest (large-scale transcriptomic), without understanding the diversity of each tree (alternative splicing specificity) nor the role that these plays in the ecosystem (biological function of splicing isoforms). Without focused studies, it is impossible to therapeutic interventions, for example by small molecules that affect splicing. This objective will be transformative in the field and indeed has in the last five years come forwards as key areas os investigation, as evidenced by the recent review on the subject that focuses on the specific novel pathways of alternative splicing and splicing factors in HCC [59].
7Alcoholic liver diseaseAlcoholic liver disease (ALD) results in fibrosis, cirrhosis, HCC, and liver failure. The role of alternative splicing in ALD is less studied; however, alternative splicing is certainly affected. Recent studies on the biochemical role of the resulting protein isoforms may be useful for identifying eventual therapeutic targets for ALD. For example, sinusoidal endothelial cells and hepatic stellate cells in case of chronic alcohol consumption have also been observed to result in an excess of EDA+ FN that accumulates in the liver, stimulating cellular inflammatory response and ALD [68].
A study investigating the splicing factor Slu7 showed the benefits of focused research to understand disease pathogenesis as well as highlighting the complexity involved in deciphering the overall balance of alternative spliced isofroms and their connection to pathology . This factor has been identified as essential for the maintenance of liver homeostasis [69]; knockdown changes hundreds of alternative splicing events, altering both inclusion and exclusion of the alternative exons, many of which are implicated in lipid metabolism, inflammation, development, and progression of fibrosis and cirrhosis. Among these the splicing factor, SRSF3 was observed to result in increased exon 4 inclusion. Inclusion of exon 4 in SRSF3 causes a premature termination codon (PTC) to be included in the final transcript, resulting in nonsense-mediated decay of the transcript [70] and hence results in lower levels of the splicing factor. Decreased levels of SRSF3, together with the associated alternative splicing events have been identified in all the liver pathologies discussed thus far and consequently would lead to the derangements identified in those studies (Fig. 3), importantly increased levels of SRSF3 also affect splicing isoforms. SLU7 and SRSF3 are up-regulated in ethanol-fed mice and in patients with alcoholic steatohepatitis [71]. Knockdown of Slu7 paradoxically considering its importance in lover homeostasis significantly reduced ethanol-induced inflammation and alcoholic liver injury in ethanol-fed mice. This in part is because it was correlated with an increase in the alternative spliced form of SRSF3 including exon 4, thereby reducing SRSF3 levels to approximately wildtype levels. [71]. A further alternative splicing variation upon Slu7 knockdown that could also play a part in the beneficial effect involved sirtuin (SIRT). Ethanol-fed mice had mildly, but significantly, higher levels of SIRT1-ΔExon8 compared with pair-fed control mice. SIRT lacking exon 8, likely results in liver damage as this isoform has a lower deacetylation activity and consequently a lower anti-inflammatory effect through the series of affected biochemical pathways [49]. Slu7 knockdown normalized SIRT1-ΔExon8 elevation in the ethanol-fed mice to the levels of control mice.
8ConclusionAlternative splicing of mRNA precursors is a versatile mechanism of gene expression regulation that accounts for a considerable proportion of proteomic complexity in higher eukaryotes. The modulation is achieved through a combinatorial interplay of positive and negative regulatory signals present in the RNA, which are recognized by complexes composed of members of the SR and hnRNP protein families. Derangements in the regulation of splicing in specific genes may be the initial trigger for many diseases. Notably, subsequent to the these derangements follows many splicing variations that are consequence and not a cause of the disease. This highlights the importance of focused studies on specific systems to identify the causative factors and use this knowledge to reverse the pathological process. This strategy has been performed in the case of some inherited diseases such as spinal muscular atrophy [72], and we believe that the information is becoming available to attempt to identify and modulate splicing events that are critical in liver diseases such as NAFLD, HCC and fibrosis in general. Overall, it seems that the changes observed in expression levels of the splicing factors per se may be the most immediate point for therapeutic intervention in liver diseases.
It is essential though to identify the dysregulated factor(s) and ensure that they are a cause and not a consequence. Identifying one master regulator of splicing that modulates of all the different isoforms may provide novel therapeutic targets to approach in early stages of the pathology. However, caution is needed, first of all to ensure proper physiological levels, that is obtaining the correct stoichiometric concentration of the factors. In addition it is important to control that this correction does not affect other processes. In fact, overexpression and downregulation of these factors, can result in disease [73]. Thus, this promising therapeutic route requires further careful investigation before reaching its clinical applications.



