



Background: Congenital hypopituitarism is an uncommon cause of neonatal cholestasis. Little is known about the effect of anterior pituitary hormone on hepatic functions.
Methods: A retrospective review of the medical charts of eight infants with congenital hypopituitarism and neonatal cholestasis was performed. The results of endocrinological investigations, eye examinations, and magnetic resonance imaging were used to classify these infants.
Results: Eight infants (4 male and 4 female; mean age, 1.7 weeks) who presented with cholestatic jaundice subsequently (mean age, 7.6 weeks) developed isolated or multiple anterior pituitary hormone deficiencies. Persistent hypoglycemia, ocular abnormalities, and microphallus were often clinical signs prompting further endocrinological and radiological investigations. Septo-optic dysplasia was prevalent, occurring in five cases. Cholestasis and hepatosplenomegaly resolved within a mean of 9.7 and 10 weeks, respectively, in the majority of cases after replacement of glucocorticoid and thyroid hormones. However, transaminase levels remained high after hormone replacement. Cortisol deficiency and hypoglycemia were noted in all cases, often following stress. Hyperlipidemia persisted in one case after the resolution of cholestasis and after corticosteroid and thyroid hormone replacement therapy. Growth hormone deficiency was not corrected due to the absence of hypoglycemia after corticosteroid hormone, an infant’s age, and/or a lack of financial resources.
Conclusions: In our series, it appears that glucocorticoid and thyroid hormones play a significant role in the resolution of cholestasis and hepatosplenomegaly. A persistently elevated transaminase level and hyperlipidemia after corticosteroid and thyroid hormone replacement may indicate the need for long-term follow-up and/or growth hormone therapy.
Neonatal jaundice is most frequently caused by indirect hyperbilirubinemia; however, direct hyperbilirubinemia is always a concern as it may be attributable to genetic, metabolic, infectious, and obstructive causes. For example, persistent neonatal jaundice can be the result of hypopituitarism. Neonatal hypopituitarism with multiple hormonal deficiencies occurs in approximately one in 53,000 newborns.1 The incidence of neonatal cholestasis associated with neonatal hypopituitarism is unknown.
MethodsFrom January 1997 to April 2006, eight infants with congenital hypopituitarism were diagnosed with neonatal cholestasis at the Department of Pediatrics, Siriraj Hospital, Mahidol University, Bangkok, Thailand. Cholestasis was diagnosed by a serum direct bilirubin level greater than 2 mg/dL or more than 15% of the total bilirubin level. Neonatal cholestasis was defined as the presence of direct hyperbilirubinemia within 3 months after birth. Extrahepatic biliary atresia, other causes of bile duct obstruction, and other metabolic liver diseases were excluded in all infants. The case histories are brieflysummarized inTable I.
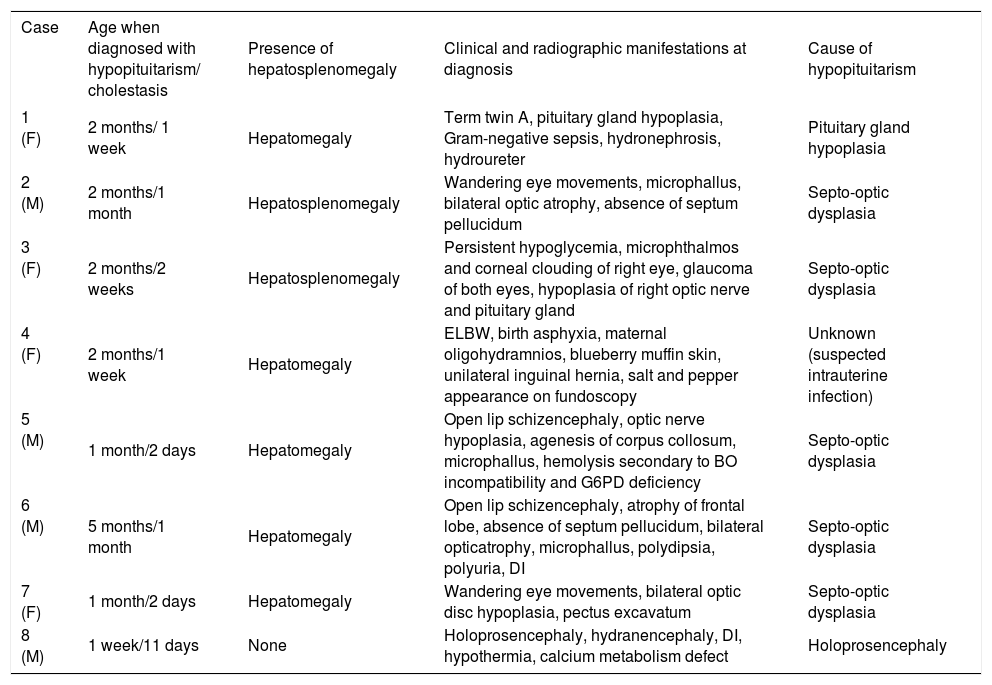
Clinical manifestations of hypopituitarism in infants in this series with neonatal cholestasis.
| Case | Age when diagnosed with hypopituitarism/ cholestasis | Presence of hepatosplenomegaly | Clinical and radiographic manifestations at diagnosis | Cause of hypopituitarism |
|---|---|---|---|---|
| 1 (F) | 2 months/ 1 week | Hepatomegaly | Term twin A, pituitary gland hypoplasia, Gram-negative sepsis, hydronephrosis, hydroureter | Pituitary gland hypoplasia |
| 2 (M) | 2 months/1 month | Hepatosplenomegaly | Wandering eye movements, microphallus, bilateral optic atrophy, absence of septum pellucidum | Septo-optic dysplasia |
| 3 (F) | 2 months/2 weeks | Hepatosplenomegaly | Persistent hypoglycemia, microphthalmos and corneal clouding of right eye, glaucoma of both eyes, hypoplasia of right optic nerve and pituitary gland | Septo-optic dysplasia |
| 4 (F) | 2 months/1 week | Hepatomegaly | ELBW, birth asphyxia, maternal oligohydramnios, blueberry muffin skin, unilateral inguinal hernia, salt and pepper appearance on fundoscopy | Unknown (suspected intrauterine infection) |
| 5 (M) | 1 month/2 days | Hepatomegaly | Open lip schizencephaly, optic nerve hypoplasia, agenesis of corpus collosum, microphallus, hemolysis secondary to BO incompatibility and G6PD deficiency | Septo-optic dysplasia |
| 6 (M) | 5 months/1 month | Hepatomegaly | Open lip schizencephaly, atrophy of frontal lobe, absence of septum pellucidum, bilateral opticatrophy, microphallus, polydipsia, polyuria, DI | Septo-optic dysplasia |
| 7 (F) | 1 month/2 days | Hepatomegaly | Wandering eye movements, bilateral optic disc hypoplasia, pectus excavatum | Septo-optic dysplasia |
| 8 (M) | 1 week/11 days | None | Holoprosencephaly, hydranencephaly, DI, hypothermia, calcium metabolism defect | Holoprosencephaly |
Congenital hypopituitarism was diagnosed by either partialorcomplete deficiency of anterior and/or posterior pituitary hormone secretion. Thyroid stimulating hormone(TSH) deficiency was indicated by low basal serum free/total thyroxin (fT4/T4) with an inappropriately normal orlow TSH. Corticotropin(ACTH)deficiency was indirectly diagnosed based on a decreased basal (8 AM) cortisol level in untreated infants and/or an inappropriately low cortisol level during hypoglycemic episodes. Growth hormone (GH) deficiency was defined as a low GH level or low insulin-like growth factor-1 (IGF-1) and/ or insulin-like binding protein-3 (IGFBP-3) level. Gonadotropin deficiency was identified as low follicle-stimulating hormone (FSH) and/or luteinizing hormone (LH) levels for a given age.
Brain magnetic resonance imaging (MRI) was performed in seven of the eight cases to identify abnormal intracranial structures. A brain imaging study was not conducted in Case 4 because of the infant’s extremely low birth weight. Septo-optic dysplasia (SOD) was identified by the presence of unilateral or bilateral optic nerve hypoplasia, which may be associated with various pituitary hormone deficiencies and central nervous system malformation.
ResultsFour male and four female infants, ranging in age from 2 days to 1 month (mean, 1.7 weeks), presented with a history of cholestatic jaundice. The following mean (range) levels were measured at the onset of cholestasis: total bilirubin, 9 mg/dL (6.5-12.3 mg/dL); direct bilirubin, 4.9 mg/dL (1.5-8.5 mg/dL); aspartate aminotransferase (AST), 210 U/L (26-666 U/L); alanine aminotransferase (ALT), 109 U/L (8-284 U/L); alkaline phosphatase, 287 U/L (237-562 U/L); gamma-glutamyl transpeptidase (GGT), 411 U/L (81-515 U/L); and albumin, 3.8 g/dL (3.3-4.2 g/dL).
Ophthalmologic examination and brain MRI revealed the cause of hypopituitarism in six of seven cases. The most common etiology of congenital hypopituitarism in our series appeared to be SOD, which was present in five of the eight cases. Case 3 exhibited microphthalmos, corneal clouding, and unilateral optic nerve hypoplasia, which represent a variant manifestation of SOD.
Serology tests for Toxoplasma gondii, rubella, herpes simplex virus (HSV), cytomegalovirus (CMV), syphilis, and urine CMV were negative in all cases and in the mothers, except in Case 2. Case 2 had positive IgG and IgM for rubella and CMV. His mother was immune to rubella and had negative IgM for T. gondii, rubella, HSV, CMV, and syphilis. His ophthalmologic exam showed optic disc hypoplasia but did not demonstrate chorioretinitis, microphthalmia, glaucoma, cataract, or other abnormalities. His liver biopsy revealed giant cell hepatitis without a viral inclusion body or cytopathic effect, as well as negative periodic acid Schiff (PAS)-stained granules in the hepatocytic cytoplasm after diastase application, which is inconsistent with alpha-1 antitrypsin deficiency. In contrast, Case 4 had negative serology tests but a clinical presentation similar to congenital infection. Testing for a mutation in the genes encoding pituitary transcription factors was not performed in any of these cases.
Persistent hypoglycemia, abnormal ocular findings, and microphallus led to endocrinological investigations in our cases with prolonged jaundice (Table I). The evaluation of anterior pituitary function was performed directly by measuring for deficiencies in TSH, GH, FSH, and LH levels and indirectly by measuring the cortisol level for ACTH deficiency and the IGF-1 and IGFBP-3 levels for GH deficiency (Table II); however, it must be considered that the normal limits for hormones are different during different periods of life and in various clinical manifestations. Hypopituitarism was diagnosed in these cases at 1-20 weeks of age (mean, 7.6 weeks). The presence of a low or low-normal serum fT4 level with a low or normal serum TSH level (Cases 1, 3-5, and 8) or, even more so, an inappropriately normal or mildly elevated serum TSH level in the presence of low-normal free T4 (Cases 2 and 6) indicated central hypothyroidism. Serum cortisol levels were low in all cases. Profoundly diminished serum cortisol levels during stress, particularly during hypoglycemic episodes, would have strongly suggested cortisol deficiency. Cortisol deficiency and hy-poglycemia were not initially present in Case 6, but appeared following stress.
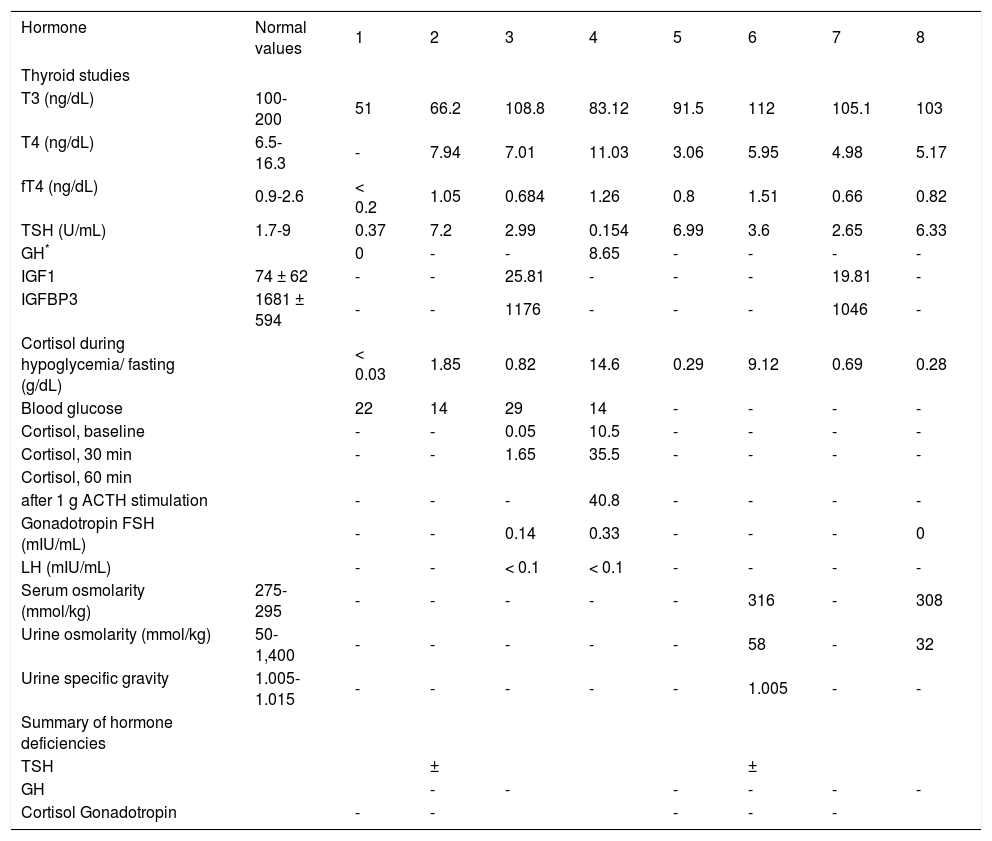
Endocrinological investigations in eight jaundiced infant cases with congenital hypopituitarism.
| Hormone | Normal values | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| Thyroid studies | |||||||||
| T3 (ng/dL) | 100-200 | 51 | 66.2 | 108.8 | 83.12 | 91.5 | 112 | 105.1 | 103 |
| T4 (ng/dL) | 6.5-16.3 | - | 7.94 | 7.01 | 11.03 | 3.06 | 5.95 | 4.98 | 5.17 |
| fT4 (ng/dL) | 0.9-2.6 | < 0.2 | 1.05 | 0.684 | 1.26 | 0.8 | 1.51 | 0.66 | 0.82 |
| TSH (U/mL) | 1.7-9 | 0.37 | 7.2 | 2.99 | 0.154 | 6.99 | 3.6 | 2.65 | 6.33 |
| GH* | 0 | - | - | 8.65 | - | - | - | - | |
| IGF1 | 74 ± 62 | - | - | 25.81 | - | - | - | 19.81 | - |
| IGFBP3 | 1681 ± 594 | - | - | 1176 | - | - | - | 1046 | - |
| Cortisol during hypoglycemia/ fasting (g/dL) | < 0.03 | 1.85 | 0.82 | 14.6 | 0.29 | 9.12 | 0.69 | 0.28 | |
| Blood glucose | 22 | 14 | 29 | 14 | - | - | - | - | |
| Cortisol, baseline | - | - | 0.05 | 10.5 | - | - | - | - | |
| Cortisol, 30 min | - | - | 1.65 | 35.5 | - | - | - | - | |
| Cortisol, 60 min | |||||||||
| after 1 g ACTH stimulation | - | - | - | 40.8 | - | - | - | - | |
| Gonadotropin FSH (mIU/mL) | - | - | 0.14 | 0.33 | - | - | - | 0 | |
| LH (mIU/mL) | - | - | < 0.1 | < 0.1 | - | - | - | - | |
| Serum osmolarity (mmol/kg) | 275-295 | - | - | - | - | - | 316 | - | 308 |
| Urine osmolarity (mmol/kg) | 50-1,400 | - | - | - | - | - | 58 | - | 32 |
| Urine specific gravity | 1.005-1.015 | - | - | - | - | - | 1.005 | - | - |
| Summary of hormone deficiencies | |||||||||
| TSH | ± | ± | |||||||
| GH | - | - | - | - | - | - | |||
| Cortisol Gonadotropin | - | - | - | - | - |
GH*, Growth hormone measured during hypoglycemic episodes. -, Not available. ±, Borderline level but probably deficient due to relatively low TSH level to fT4 level.
Low IGF-1, IGFBP-3, and/or GH levels in the presence of hypoglycemia (Cases 1, 3, 4 and 7) were interpreted as GH deficiency. Gonadotropins were measured in only three patients (Cases 3, 4, and 8). The data available from our patients demonstrated low gonadotropin levels. Cases 6 and 8 developed central diabetes insipidus (DI).
After the diagnosis of anterior pituitary hormone deficiency, hormone replacement therapy (HRT) was begun. GH therapy was not done due to the absence of hypoglycemia after corticosteroid hormone, an infant’s age, and/ or a lack of financial resources. Levothyroxine was administered at a dose of ~50 μg/m2 per day for the treatment of central hypothyroidism. Oral hydrocortisone was given two or three times daily at a dose of 10-15 mg/m2 per day. Treatment was initiated after identifying deficient hormones. In Cases 2 and 3, cholestasis improved and ultimately resolved after the administration of only glucocorticoid hormone, whereas both thyroid and glucocorticoid hormones were required to improve cholestasis in Cases 1, 4, and 7. Cholestasis and hepatosplenomegaly resolved within a mean of 9.7 and 10 weeks, respectively, after the replacement of glucocorticoid and/or thyroid hormones in Cases 1-5 and 7. In Case 6, the resolution of cholestasis required the addition of ursodeoxycholic acid to the hormone replacement treatment. Liver dysfunction spontaneously improved in Case 8. The elevated transaminase level in Case 4 was improved after 6 weeks, whereas it persisted for more than 6 weeks in Cases 1 and 7, despite the replacement of glucocorticoid and thyroid hormones.
Hyperlipidemia was observed in Case 4, with fasting blood levels of 49 mg/dL HDL-C, 128 mg/dL LDL-C, 251 mg/dL cholesterol, and 408 mg/dL triglyceride, despite the resolution of cholestasis. Total parenteral nutrition was administered for 7 days but did not change the presence of hyperlipidemia in the absence of cholestasis. No familial hyperlipidemia was noted in this case.
DiscussionIn adults, pituitary adenoma or its associated therapy with surgery and radiation is the most common cause of hypopituitarism.2 Anatomical anomalies associated with SOD, such as anencephaly, holoprosencephaly, hydrocephalus, and midline defects; birth trauma and/or asphyxia; mutations in genes encoding pituitary transcription factors; and congenital or intrauterine infections have been reported in infants with congenital hypopituitarism.3-5 A brain MRI scan is essential for establishing these pathologies. The majority of infants with congenital hypopituitarism exhibit multiple pituitary hormone deficiencies.6 Hypothetically, the disruption of the hypothalamic pituitary axis could originate from mutations in any of the genes encoding critical axis components, such as the PROP 1 and Pit 1 transcription factors, leading to multiple pituitary hormone deficiencies.7
The etiology of SOD is multi-factorial and includes viral infection, gestational diabetes, vascular disruption, and genetic mutations. The HESX1 gene, which encodes a homeobox gene expressed in embryonic stem cells, plays an important role in the development of the optic nerves as well as the anterior pituitary gland. Its mRNA and protein can be found in many organs, including the liver and brain. Mutation of the HESX1 gene has been identified in SOD.7 Sixty percent of patients with SOD manifest pituitary gland dysfunction secondary to decreased levels of hypothalamic releasing factors.8 Up to 93% of patients with optic nerve hypoplasia have GH deficiency, which is the most common pituitary hormone deficiency. Complex microphthalmos and corneal clouding have also been observed in a SOD variant,9 as seen in Case 3 of the present series. In this situation, the diagnosis of optic nerve and chiasmal hypoplasia can be made by MRI.
The full-term infant has immature hepatic metabolism and biliary excretion. Bile flow is characterized by two mechanisms of biliary excretion, bile acid-dependent and bile acid-independent flow. Bile acid-dependent flow involves active water flow and the diffusion of solutes,10 whereas bile acid-independent flow is driven by active transporters of anions and cations, such as Na+, K+-adenosine triphosphatase (ATPase).
The etiology of neonatal cholestasis consists of numerous causes, which can be isolated or multifactorial. Cholestasis in hypopituitarism insidiously occurs at birth, together with or following physiological jaundice. Ellaway et al. reported that seven of 20 children with congenital hypopituitarism exhibited cholestatic jaundice as a major presentation.11 The pathogenic mechanism of the development of cholestasis in hypopituitarism is unclear. Layden and Boyer reported that thyroid hormone and cortisol affect the bile acid-independent bile flow.12 Isolated thyroid hormone, TSH, GH, ACTH, and cortisol deficiencies have been shown to cause conjugated hyperbilirubinemia, but combined deficiencies have also been described.13,14
Experiments have demonstrated that cortisol can influence bile formation; reduced bile flow has been observed in rats after adrenalectomy.15 Hydrocortisone infusion has been shown to increase bile acid-independent bile flow in the dog and rat.16,17 Layden and Boyer reported canalicular bile formation, a complex process dependent on the osmotic effects of bile salt excretion as well as bile acid-independent flow.12 In thyroidectomized rats, thyroid hormone altered bile secretory function by influencing both the flow rate and amount of bile.18 Thyroid hormone has a similar effect on bile acid-independent canalicular secretion and the liver plasma membrane Na+, K+-ATPase activity. Although cortisol and thyroid hormones promote bile acid-independent flow, there is little contribution of the bile acid-independent component during the neonatal period.19
GH has been shown to modulate bile acid synthesis in both rats and humans.20,21 Brewer demonstrated that GH can modulate bile acid synthesis.12 Giacoia et al. suggested that GH was necessary for bile acid secretion rather than synthesis.13 Poley et al. studied the composition of the micellar phase during the digestion of two consecutive meals in GH-deficient and control patients before and after HRT with GH replacement.21 They concluded that HRT with GH increased the hepatobiliary secretion of bile acids and postulated that GH influences hepatic bile acid biosynthesis.
The fact that jaundice improves after HRT suggests that a deficiency of pituitary hormones may be related to the pathogenesis of cholestasis. Sheehan et al. claimed that the deficiency of one or more hormones in patients with pituitary hypofunction could either delay the normal maturation of hepatic active transport mechanisms or inhibit bile acid synthesis, thereby promoting the accumulation of bile acid precursor and producing a cholestatic effect.22 This proposal agrees with the theory that hormone deficiencies can cause abnormalities of biliary structure and of the function of bile canaliculi essential for bile excretion, beyond the effects of the immature hepatobiliary system in neonates.
Based on their finding that HRT with GH increased the hepatobiliary secretion of bile acids, Poley et al. postulated that GH influenced hepatic bile acid biosynthesis.23 GH deficiency in neonatal cholestasis with congenital hypopituitarism has been documented as an etiological factor of persistent liver dysfunction in several cases receiving adequate dosages of hydrocortisone.20,21 This may explain the elevated transaminase levels that persisted in Cases 1 and 7 even after sufficient dosages of glucocorticoid and thyroid hormones were given. Theoretically, elevated transaminase levels could play a role in the development of chronic liver disease if the GH deficiency were not effectively treated.
A choleretic response to a specific anterior pituitary hormone has not been clinically well described.11 Hodges and Buckler reported that cholestasis disappeared in an infant within 1 month after GH and hydrocortisone replacement but that the elevated transaminase level did not resolve until 7 months later.21 In our series, cholestasis improved and ultimately resolved after the administration of thyroid and/or glucocorticoid hormones in most of the cases. Interestingly, spontaneous improvement of liver dysfunction was noted in one of our cases, as has been reported by Leblanc et al.14 In addition, some children who developed neonatal cholestasis were diagnosed at a later time with GH deficiency. One reported SOD case was diagnosed with GH and ACTH deficiencies at the age of 9 months, and TSH deficiency did not develop until 18 months later.24
Hypopituitarism is associated with hyperlipidemia, but the mechanism is unclear. Studies on the pathogenesis of hyperlipidemia found that plasma lipoprotein lipase and hepatic triglyceride lipase activities were suboptimal in patients with hypothyroidism.25 The fact that hyperlipidemia subsided after corticosteroid and thyroid hormone replacement suggests that the reduced activities of these two lipases may, at least in part, account for the development of hyperlipidemia in hypopituitarism. In another study, none of the GH-deficient adult patients with hypopituitarism received GH replacement but nevertheless exhibited moderate improvement after corticosteroid and thyroid hormone replacement. Increased cardiovascular mortality and high incidence of coronary disease are also associated with adult hypopituitarism.26
Hepatomegaly was present in seven of the eight cases and resolved with hormone replacement, as has been described by others.14 Splenomegaly, which is not commonly recognized in neonatal cholestasis with hypopituitarism, has also been shown to disappear after hormone therapy.14 The pathogenesis of hepatosplenomegaly is still unclear, but it is thought to be related to hematopoiesis.27 The disappearance of hepatosplenomegaly and an improved liver condition have also been reported in response to treatment with glucocorticoid hormone.14 There was no laboratory or pathological evidence of any infectious or infiltrative systemic disease that could have explained the presence of hepatosplenomegaly.14 Hepatomegaly is often correlated with abnormal liver function test results.28,29
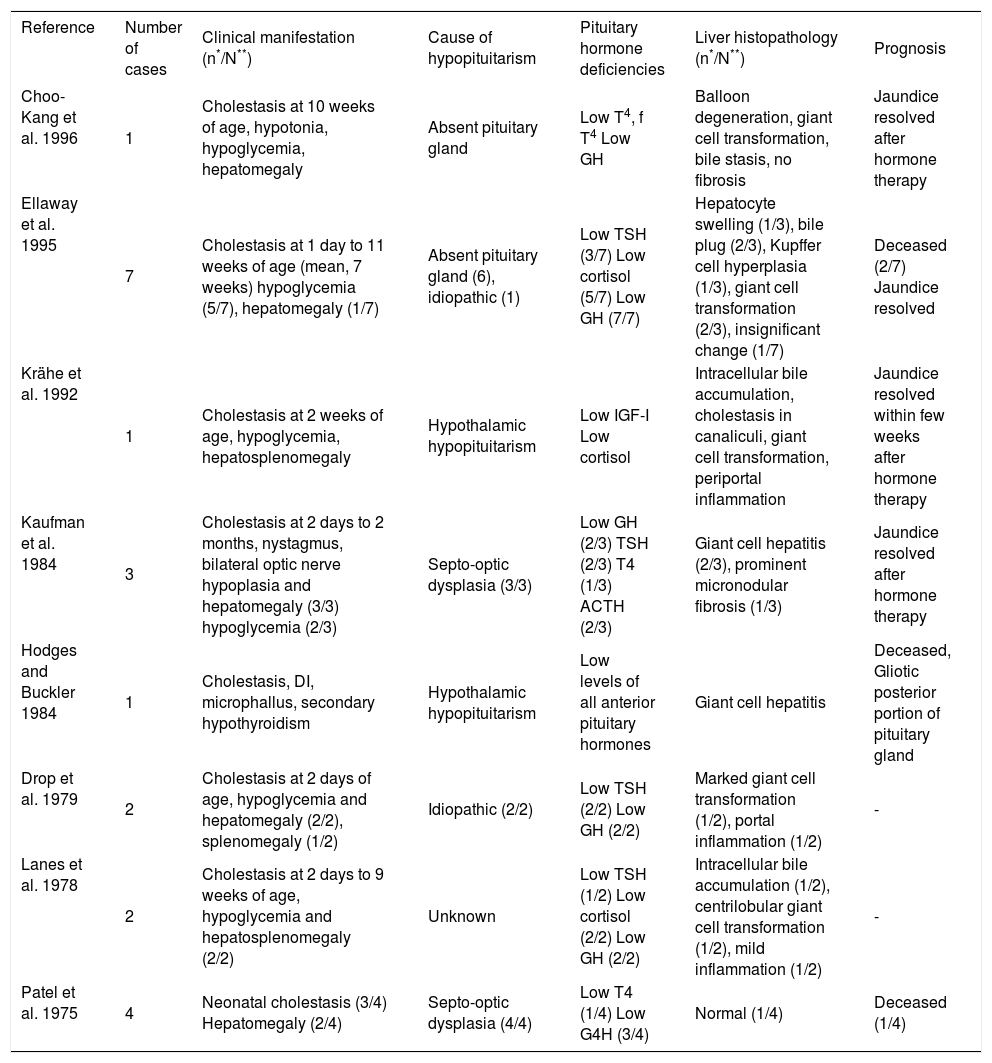
Table III summarizes the liver histopathology and outcomes for cases reported previously in the literature. These reports demonstrated inflammatory infiltration of periportal areas, bile plugs, giant cell hepatitis, intrahepatic bile duct hypoplasia, and Kupffer cell hyperplasia.1,21,23,30 Three patients showed hepatic fibrosis or cirrhosis on liver biopsies and at post mortem examination.29,31 Minami et al. reported a 7-year-old girl with SOD who presented with hepatosplenomegaly but never exhibited persistent neonatal jaundice; an abdominal ultrasonogram revealed an enlarged liver with increased echogenicity, prompting a percutaneous liver biopsy.32 The finding was consistent with congenital hepatic fibrosis, which is likely to be a coincidental finding. Kaufman et al. described persistent hepatomegaly and distortion of the lobular architecture attributable to prominent micronodular fibrosis on a repeat liver biopsy in a patient with SOD.31 The data from liver biopsies generally give neither a specific diagnosis nor prognosis of the disease. Given the availability of other noninvasive methods for determining etiology, percutaneous liver biopsy should be considered as a last resort for patients in whom hypopituitarism is the probable etiology of neonatal cholestasis.
Hepatic manifestations in hypopituitarism: Clinical, biochemical, and histopathological characteristics reported previously in infants with cholestasis.
| Reference | Number of cases | Clinical manifestation (n*/N**) | Cause of hypopituitarism | Pituitary hormone deficiencies | Liver histopathology (n*/N**) | Prognosis |
|---|---|---|---|---|---|---|
| Choo-Kang et al. 1996 | 1 | Cholestasis at 10 weeks of age, hypotonia, hypoglycemia, hepatomegaly | Absent pituitary gland | Low T4, f T4 Low GH | Balloon degeneration, giant cell transformation, bile stasis, no fibrosis | Jaundice resolved after hormone therapy |
| Ellaway et al. 1995 | 7 | Cholestasis at 1 day to 11 weeks of age (mean, 7 weeks) hypoglycemia (5/7), hepatomegaly (1/7) | Absent pituitary gland (6), idiopathic (1) | Low TSH (3/7) Low cortisol (5/7) Low GH (7/7) | Hepatocyte swelling (1/3), bile plug (2/3), Kupffer cell hyperplasia (1/3), giant cell transformation (2/3), insignificant change (1/7) | Deceased (2/7) Jaundice resolved |
| Krähe et al. 1992 | 1 | Cholestasis at 2 weeks of age, hypoglycemia, hepatosplenomegaly | Hypothalamic hypopituitarism | Low IGF-I Low cortisol | Intracellular bile accumulation, cholestasis in canaliculi, giant cell transformation, periportal inflammation | Jaundice resolved within few weeks after hormone therapy |
| Kaufman et al. 1984 | 3 | Cholestasis at 2 days to 2 months, nystagmus, bilateral optic nerve hypoplasia and hepatomegaly (3/3) hypoglycemia (2/3) | Septo-optic dysplasia (3/3) | Low GH (2/3) TSH (2/3) T4 (1/3) ACTH (2/3) | Giant cell hepatitis (2/3), prominent micronodular fibrosis (1/3) | Jaundice resolved after hormone therapy |
| Hodges and Buckler 1984 | 1 | Cholestasis, DI, microphallus, secondary hypothyroidism | Hypothalamic hypopituitarism | Low levels of all anterior pituitary hormones | Giant cell hepatitis | Deceased, Gliotic posterior portion of pituitary gland |
| Drop et al. 1979 | 2 | Cholestasis at 2 days of age, hypoglycemia and hepatomegaly (2/2), splenomegaly (1/2) | Idiopathic (2/2) | Low TSH (2/2) Low GH (2/2) | Marked giant cell transformation (1/2), portal inflammation (1/2) | - |
| Lanes et al. 1978 | 2 | Cholestasis at 2 days to 9 weeks of age, hypoglycemia and hepatosplenomegaly (2/2) | Unknown | Low TSH (1/2) Low cortisol (2/2) Low GH (2/2) | Intracellular bile accumulation (1/2), centrilobular giant cell transformation (1/2), mild inflammation (1/2) | - |
| Patel et al. 1975 | 4 | Neonatal cholestasis (3/4) Hepatomegaly (2/4) | Septo-optic dysplasia (4/4) | Low T4 (1/4) Low G4H (3/4) | Normal (1/4) | Deceased (1/4) |
n*/N**, Number of cases with manifestation(s) per total number of cases.
The unique features of congenital hypopituitarism which can assist in its diagnosis include persistent hypoglycemia, ocular manifestations, microphallus, and neonatal cholestasis. Cortisol deficiency and hypoglycemia may not present initially but may appear following stress, when compensation can no longer occur. The unknown long-term effects of hypopituitarism on hepatic function and hyperlipidemia necessitate continued clinical observations and, possibly, adequate GH therapy.
Acknowledgement:The authors also wish to thank Professor Chanika Tuchinda, Professor Kitti Angsusingha, and Professor Chirasri Vajaradul, who contributed the care and investigations of these patients.



