



Progressive familial intrahepatic cholestasis type 3 (PFIC-3) is a rare autosomal recessive cholestatic liver disorder caused by mutations in the ABCB4 gene. The aim of this study was to present the phenotypic and genotypic spectrum of 4 Polish PFIC-3 patients diagnosed in a one-referral centre.
Materials and methodsThe study included 4 patients with cholestasis and pathogenic variants in the ABCB4 gene identified by next-generation sequencing (NGS) of a targeted-gene panel or whole exome sequencing (WES). Clinical, laboratory, histological, and molecular data were collected.
ResultsFour patients (three males) were identified. The age at first noted clinical signs and symptoms was 6, 2.5, 14, and 2 years respectively; the mean age was 6 years. Those signs and symptoms include pruritus (2 out of 4 patients) and hepatomegaly with splenomegaly (4 out of 4 patients). The age at the time of referral to our centre was 9, 3, 15, and 2.5 years respectively, while the mean age was 7 years. Chronic cholestatic liver disease of unknown aetiology was established in all of them. The NGS analysis was performed in all patients at the last follow-up visit. Three novel variants including c.902T>A, p.Met301Lys, c.3279+1G>A, p.?, and c.3524T>A, p.Leu1175His were identified. The time from the first consultation to the final diagnosis was 14, 9, 3, and 1 year respectively; the mean was 6.8 years. A detailed follow-up was presented.
ConclusionsThe clinical phenotype of PFIC-3 could be variable. The clinical and biochemical diagnosis of PFIC-3 is difficult, thus the NGS study is very useful in making a proper diagnosis.
Progressive familial intrahepatic cholestasis type 3 (PFIC-3, # 602347) is a rare autosomal recessive cholestatic liver disorder caused by mutations in the adenosine triphosphate-binding cassette subfamily B member 4 (ABCB4) gene, encoding the human multidrug resistance 3 (MDR3) protein [1–3]. MDR3 is expressed on the canalicular membrane of the hepatocyte and is responsible for the efflux of phosphatidylcholine (PC), playing an important role in the protection of hepatocytes and cholangiocytes from the detergent action of free bile acids (BA) [1–3].
PFIC-3 patients are usually homozygous or compound heterozygous for ABCB4 pathogenic variants; however, monoallelic ABCB4 variants may also result in cholestatic liver disease [4–6]. Monoallelic or biallelic ABCB4 variants have also been described in adult low phospholipid-associated cholelithiasis (LPAC, # 600803), intrahepatic cholestasis of pregnancy 3 (ICP3; # 614972), and drug-induced liver disease (DILI) [7–10]. Due to the clinical and molecular heterogeneity of resulting phenotypes (PFIC-3, LPAC, ICP3, DILI), there is a need for research aimed at a better characterisation of these peculiar diseases.
The aim of this study was to present the phenotypic and genotypic spectrum of 4 Polish PFIC-3 patients diagnosed in a one-referral centre based on an NGS study to highlight diagnostic difficulties, find 3 novel ABCB4 variants, and also to provide the long-term follow-up.
2Material and methodsPatients with chronic intrahepatic cholestasis of unknown cause were enrolled in a single paediatric referral centre. Next-generation sequencing (NGS) of a targeted-gene panel, created by the Children's Memorial Health Institute for the simultaneous sequencing of 1000 clinically relevant genes including 54 items related to cholestatic liver disorders or cholestasis as syndromic features, was used for Patients 2, 3, and 4, whereas whole exome sequencing was applied to Patient 1. A detailed study protocol has been described recently [11,28].
The nomenclature of molecular variants follows the Human Genome Variation Society guidelines (HGVS, http://varnomen.hgvs.org/) using a human cDNA sequence of the ABCB4 gene followed the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk).
The study included 4 patients with cholestasis and pathogenic variants in ABCB4.
Clinical, laboratory, histological, and molecular data were collected.
An informed and written consent was obtained from patients and their parents. An ethical approval of the study protocol was obtained from the Bioethical Committee of the Children's Memorial Health Institute, Warsaw, Poland.
3Results3.1Presentation at time of referralFour patients (three males) were identified. The age at first noted clinical signs and symptoms was as follows: 6 years (Patient 1), 2.5 years (Patient 2), 14 years (Patient 3), and 2 years (Patient 4); the mean age was 6 years. Those signs and symptoms include pruritus (Patients 2 and 4) and hepatosplenomegaly (all patients).
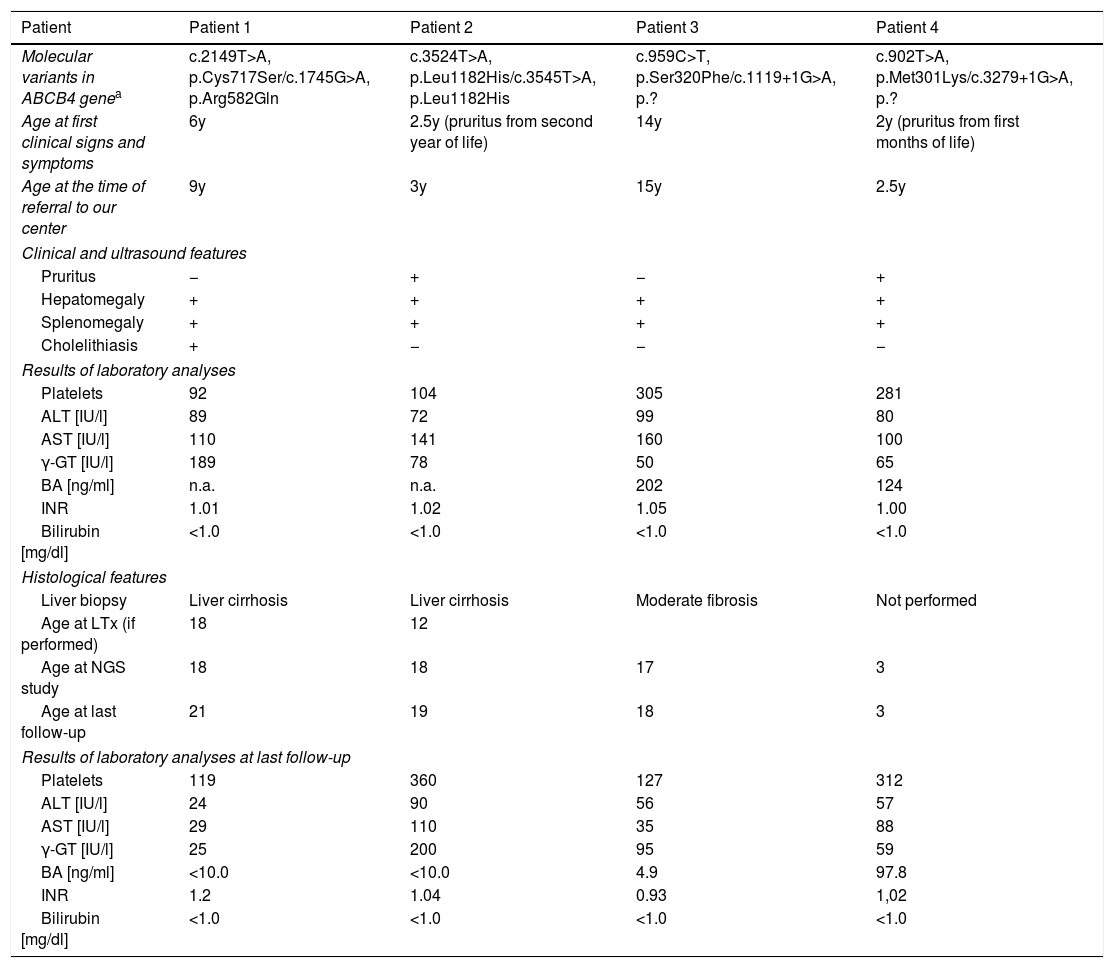
The age at the time of referral to our centre was 9 years (Patient 1), 3 years (Patient 2), 15 years (Patient 3), and 2.5 years (Patient 4); the mean age was 7 years. Presenting features included hepatomegaly (all patients), splenomegaly (all patients), pruritus (Patients 2 and 4), and cholelithiasis (Patient 2). In all patients, laboratory analyses revealed the presence of elevated serum transaminases, elevated GGT activity, elevated serum BA concentration, normal serum total and direct bilirubin concentration, and no coagulopathy (normal INR), with thrombocytopenia being noted in two patients (Patients 1 and 2). As for the serum concentration of vitamins A, E and D, only vitamin D was deficient in three patients at the time of referral. Clinical and biochemical features are presented in Table 1.
Detailed characteristics on the study group.
| Patient | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Molecular variants in ABCB4 genea | c.2149T>A, p.Cys717Ser/c.1745G>A, p.Arg582Gln | c.3524T>A, p.Leu1182His/c.3545T>A, p.Leu1182His | c.959C>T, p.Ser320Phe/c.1119+1G>A, p.? | c.902T>A, p.Met301Lys/c.3279+1G>A, p.? |
| Age at first clinical signs and symptoms | 6y | 2.5y (pruritus from second year of life) | 14y | 2y (pruritus from first months of life) |
| Age at the time of referral to our center | 9y | 3y | 15y | 2.5y |
| Clinical and ultrasound features | ||||
| Pruritus | − | + | − | + |
| Hepatomegaly | + | + | + | + |
| Splenomegaly | + | + | + | + |
| Cholelithiasis | + | − | − | − |
| Results of laboratory analyses | ||||
| Platelets | 92 | 104 | 305 | 281 |
| ALT [IU/l] | 89 | 72 | 99 | 80 |
| AST [IU/l] | 110 | 141 | 160 | 100 |
| γ-GT [IU/l] | 189 | 78 | 50 | 65 |
| BA [ng/ml] | n.a. | n.a. | 202 | 124 |
| INR | 1.01 | 1.02 | 1.05 | 1.00 |
| Bilirubin [mg/dl] | <1.0 | <1.0 | <1.0 | <1.0 |
| Histological features | ||||
| Liver biopsy | Liver cirrhosis | Liver cirrhosis | Moderate fibrosis | Not performed |
| Age at LTx (if performed) | 18 | 12 | ||
| Age at NGS study | 18 | 18 | 17 | 3 |
| Age at last follow-up | 21 | 19 | 18 | 3 |
| Results of laboratory analyses at last follow-up | ||||
| Platelets | 119 | 360 | 127 | 312 |
| ALT [IU/l] | 24 | 90 | 56 | 57 |
| AST [IU/l] | 29 | 110 | 35 | 88 |
| γ-GT [IU/l] | 25 | 200 | 95 | 59 |
| BA [ng/ml] | <10.0 | <10.0 | 4.9 | 97.8 |
| INR | 1.2 | 1.04 | 0.93 | 1,02 |
| Bilirubin [mg/dl] | <1.0 | <1.0 | <1.0 | <1.0 |
The nomenclature of identified variants follows the Human Genome Variation Society guidelines (HGVS v 2.0, www.hgvs.org/mutnomen) and referral to the cDNA and protein sequences: NM_000443.3, NP_000434.1 for ABCB4; followed the Human Gene Mutation Database (HGMD, www.hgmd.cf.ac.uk). Boldface print – unpublished/novel variants.
The peculiar diagnostic process of cholestatic liver disorders was introduced as described in our previous report [11].
Liver biopsies were done in three patients (Patients 1, 2, 3) at the age of 11, 5, and 16 years respectively. The presence of chronic hepatitis of mild activity (grade 2 according to the Batts and Ludwig classification) with inflammatory infiltrates composed of neutrophils and lymphocytes, and bridging fibrosis (stage 3) was observed in Patient 1 at the age of 11 years (5 years after the first signs and symptoms). Patient 2 was diagnosed with liver cirrhosis (stage 4) and chronic hepatitis with mild activity (grade 2) at the age of 5 years (2.5 years after the first signs and symptoms). Moderate liver fibrosis reflecting stage 2 according to the Batts and Ludwig classification was identified in Patient 3 at the age of 16 years (1 year after the first signs and symptoms).
3.2Presentation during follow-upThe follow-up was as follows: 12 years (Patient 1), 16 years (Patient 2), 3 years (Patient 3), and 1 year (Patient 4).
Progressive thrombocytopenia, cholestatic jaundice, and several focal liver lesions with slightly elevated AFP levels were observed in Patient 1 from the age of 16. Another liver biopsy was performed at the age of 16, which revealed the presence of cirrhosis (stage 4) and chronic hepatitis with moderate activity (grade 3). At the age of 18, he was qualified for LTx, presenting with massive splenomegaly, cholelithiasis, and cholestatic jaundice with a history of several episodes of gastroesophageal variceal bleeding. Laboratory results before LTx were as follows: leukocytes 3×103/μl, platelets 28×103/μl, AST 134IU/L, ALT 61IU/L, GGT 391IU/L, total/direct serum bilirubin 5.4/3.8mg/dl, INR 1.15, AFP 30ng/ml. Liver transplantation was performed at the age of 19, with hepatocellular carcinoma (HCC) being diagnosed in the explanted liver. At the last follow-up (21 years), the patient presented with normal liver function (Table 1) on tacrolimus-based immunosuppressive treatment.
At the follow-up, Patient 2 presented with massive splenomegaly, progressive thrombocytopenia, and leukopenia. Laboratory analyses at the age of 11 were as follows: leukocytes 1.8×103/μl, platelets 18×103/μl, AST 81IU/L, ALT 31IU/L, GGT 112IU/L, total/direct serum bilirubin 4.1/3.6mg/dl, INR 1.6, albumin 3.2g/dl, AFP 2.4IU/ml. Gastroscopy performed at that age revealed the presence of oesophageal varices (grade I). Progressive cholestatic jaundice and coagulopathy (prolonged INR) were observed from 11 years of age. She underwent liver transplantation (LTx) with splenectomy at 12 due to liver cirrhosis of unknown aetiology. At the last follow-up (19 years), the patient presented with normal liver function (Table 1) on tacrolimus-based immunosuppressive treatment.
Patient 3, at 18 years of age (2 years after the referral), presented in a good clinical condition with moderate splenomegaly (the liver not palpable below the rib arch), no jaundice, normal AST activity, elevated ALT (168IU/L) and GGT (95IU/L) activity, and normal serum BA concentration on UDCA treatment (Table 1). Gastroscopy was normal.
Patient 4, at 3.5 years of age (1 year after the referral), presented in a good clinical condition with splenohepatomegaly, no jaundice, elevated AST (88IU/L), ALT (46IU/L) and GGT (90IU/L) activity, and elevated serum BA concentration (98μmol/l) despite UDCA treatment (Table 1). Gastroscopy was planned for the next visit.
3.3Molecular analysis resultsThe NGS analysis was performed in all patients at the last follow-up visit (2018–2019); seven variants in the ABCB4 gene (RefSeq NM_000443.3) were identified in the study group. Three variants including c.902T>A, p.Met301Lys, c.3279+1G>A, p.?, and c.3524T>A, p.Leu1175His were novel (Table 1). Four other variants, c.959C>T, p.Ser320Phe, c.1119+1G>A, p.?, c.1745G>A, p.Arg582Gln, c.2149T>A, p.Cys717Ser were described previously, but one of them (c.1119+1G>A) was identified to date only in a Polish patient [11].
The age at PFIC-3 diagnosis was as follows: 18 years (Patient 1), 18 years (Patient 2), 17 years (Patient 3), and 3 years (Patient 4).
The time from the first consultation to the final diagnosis was as follows: 14 years (Patient 1), 9 years (Patient 2), 3 years (Patient 3), and 1 year (Patient 4); the mean time was 6.8 years.
4DiscussionThis study provides a comprehensive clinical, biochemical, histological, and molecular description of additional four PFIC-3 patients.
Progressive familial intrahepatic cholestasis is a collectively used term for several diseases with a discreet molecular background, among which PFIC-3 constitutes a peculiar disease [2]. Unlike PFIC-1 (FIC1 deficiency), PFIC-2 (BSEP deficiency) and PFIC-4 (TJP2 deficiency), PFIC-5 (FXR deficiency) and MYO5B deficiency, PFIC-3 (MDR3 deficiency) patients rarely present with neonatal cholestatic jaundice; this more often occurs in late infancy, childhood, or even adulthood. Therefore, it is difficult to suspect this type of disease referred to as “cholestasis” in children who do not present with jaundice [5,12]. In a cohort of 427 patients with suspected genetic cholestasis, Dröge et al. (2017) observed that the median age of the onset of symptoms in PFIC-3 patients was later than in PFIC-1 or PFIC-2 patients [13]. This is well reflected in our study results, where the age of the patient at first clinical signs and symptoms was 2.5–14 years (mean 6 years). There is a need for careful and systematic monitoring of these patients as observed during the follow-up of Patients 1 and 2, who at first presented with hepatosplenomegaly and developed cholestatic jaundice and features of portal hypertension later in the course of the disease (7 and 8 years after the referral respectively). The primary defect in MDR3 deficiency does not cause retention of bile acids in the hepatocyte and, therefore, does not directly cause cholestasis [14]. Symptoms occur as a consequence of cholangiocyte damage. MDR3 is a phospholipid translocator involved in biliary phospholipid (phosphatidylcholine) excretion; therefore, the absence of phospholipids in bile destabilises micelles and promotes bile lithogenicity with cholesterol crystallisation.
This reflects the main difference in PFIC-3 pathophysiology, distinguishing it from other PFIC disorders.
It is common knowledge that serum γ-GT activity is normal in PFIC-1 and PFIC-2 patients but is elevated in PFIC-3 patients [15]. In the study by Colombo et al., a total of 28 PFIC-3 patients were diagnosed among 133 children with chronic intrahepatic cholestasis with elevated GGT activity [16]. Interestingly, eight out of these 28 patients were asymptomatic, with PFIC-3 being incidentally discovered through abnormalities in liver enzymes.
Recently, Schatz et al. described national survey data on PFIC-3, highlighting that the earlier the disease onset, the more severe phenotype could be observed and patients with milder phenotypes could be not diagnosed before adulthood [17]. All (26) patients diagnosed in childhood developed pruritus (median age 1 year), while splenomegaly and hepatomegaly were the most common first clinical signs and symptoms. Gallstones at any time were detected in only 4 patients (15%). Our results are very similar to the results of Schatz et al. and other studies in the literature. PFIC-3 patients usually present with hepatosplenomegaly, mild pruritus, and a persistent elevation of GGT activity but without jaundice [1–3]. Fluctuating activities of serum transaminases and even normal GGT activity or bilirubin concentration may contribute to the delay in correct diagnosis [17]. PFIC-3 patients have a higher risk of developing portal hypertension and gastrointestinal bleeding in young adulthood [5]. Some authors also suggest that cryptogenic cholestasis in adults should be added to the spectrum of conditions associated with ABCB4 mutations [18,19].
Liver histology in PFIC-3 is characterised by a non-specific portal inflammation, extensive portal fibrosis, cholestasis with ductular proliferation, and the loss of MDR3 protein expression [4,13,20]. In our study, the liver biopsy of Patient 1 was miscellaneous because of the presence of chronic hepatitis and only minimal cholestasis. Two patients developed liver cirrhosis. The literature includes several studies on molecular analysis for patients with cryptogenic cirrhosis, which leads to the detection of ABCB4 mutations [21].
NGS is very useful in MDR3 deficiency diagnosis due to its heterogeneous clinical presentation. Since its introduction, the time to diagnosis has decreased. It is clearly illustrated by the report on Patient 4. He was the latest patient to be diagnosed with PFIC-3 in our institute. The time from referral to diagnosis was about 1 year. In addition, the liver biopsy was not performed in the diagnostic process because of the ongoing NGS study.
258 likely pathogenic ABCB4 variants in the Human Gene Mutation Database (HGMD Professional 2020.3) have been reported so far [22]. Causative mutations include small or gross deletions/insertions, missense and nonsense mutations. A genotype-phenotype correlation is observed. Patients with residual MDR3 expression (and residual transport activity), especially those with missense mutations (like p.Ser320Phe in our study), usually present with a slowly progressive disease and could respond well to treatment with UDCA [4,5,17,23]. On the other hand, there are reports of heterozygotes for complete loss of function variants, which could induce liver injury leading to significant fibrosis [6,24]. In our study, we identified three novel ABCB4 variants, including two missense and one splice site pathogenic variant. Patient 2 was a homozygote for the p. Leu1175His variant; the PolyPhen-2 prediction has not reported this variant as probably damaging in public databases (gnomAD, Exome Variant Server). This corresponds with clinical observations; this patient was diagnosed with liver cirrhosis at 5 years of age and transplanted at 12 years of age due to end-stage liver disease. Both remaining novel variants – c.902T>A, p.Met301Lys and c.3279+1G>A, p.? – have not been reported in public databases (gnomAD, Exome Variant Server) and have been predicted as possibly pathogenic using in silico tools (CADD, PolyPhen2 HDIV, Mutation Assessor, Mutation Taster, FATHMM, LRT, MetaSVM, MetaLR for the missense variant and CADD, Mutation Taster for the splice site variant).
Patient 1 was a compound heterozygote for two missense variants: p.Cys717Ser and p.Arg582Gln. Both variants were predicted by PolyPhen-2 as possibly damaging with a score of 0.678 and 0.956 respectively. The p.Ser320Phe variant found in Patient 3 was reported in low phospholipid-associated cholelithiasis and progressive familial intrahepatic cholestasis 3 as well.
At present, liver transplantation (LTx) is the only effective treatment for end-stage liver diseases in the course of PFIC-3 [1–3,25]. The study by Schatz et al. emphasises that severe and early manifestation of PFIC-3 leads to the necessity of liver transplantation in almost 50% of patients [17]. Our study also confirms that the younger age at presentation, the more severe PFIC-3 phenotype with the need for early LTx.
Similar to BSEP deficiency, MDR3 deficiency patients have an increased risk of developing hepatocarcinoma (HCC) and cholangiocarcinoma [26,27]. In our study, HCC was diagnosed in one patient – at the age of 18 (12 years after referral to our Institute). This observation and other literature observations suggest a need for a careful and prospective follow-up of patients with ABCB4 mutations in order to identify the HCC and CCA risk.
5Conclusions- 1.
The clinical phenotype of PFIC-3 could be variable. It was observed that the younger age at presentation, the more severe PFIC-3 phenotype with the need for early LTx.
- 2.
The diagnosis of PFIC-3 is difficult because some PFIC-3 patients initially presented with no jaundice. Next-generation sequencing was very useful in final PFIC-3 diagnosis.
- 3.
PFIC-3 patients are at increased risk of HCC development thus there is a need of careful and systematic long-term monitoring of these patients.
- 4.
Three novel ABCB4 variants were identified, which included c.902T>A, p.Met301Lys, c.3279+1G>A, p.?, and c.3524T>A, p. Leu1175His. They were classified as pathogenic according to in silico analysis and our clinical observations.
adenosine triphosphate-binding cassette subfamily B member 4 gene
AFPalpha-fetoprotein
ALTalanine aminotransferase
ASTaspartate aminotransferase
BAbile acids
BSEPbile salt export pump
CCAcholangiocarcinoma
DILIdrug-induced liver disease
FXRfarnesoid X receptor
GGTgamma-glutamyl transferase
HCChepatocellular carcinoma
ICP3intrahepatic cholestasis of pregnancy 3
LPAClow phospholipid-associated cholelithiasis
LTxliver transplantation
MDR3multidrug resistance 3 protein
MYO5Bmyosin VB
NGSnext-generation sequencing
PCphosphatidylcholine
PFICprogressive familial intrahepatic cholestasis
PFIC-3progressive familial intrahepatic cholestasis type 3
TJP-2tight junction protein 2
UDCAursodeoxycholic acid
WESwhole exome sequencing
FundingThe study was partially funded by the Children's Memorial Health Institute intramural grant M28/17.
Authors’ contributionsConception and design of study: PL and IJ. Analysis and/or interpretation of data: PL, EC, DJ, RP, MW-S, JP, and IJ. Drafting the manuscript: PL, EC, and DJ. Revising the manuscript: JP and IJ.
Conflict of interestAll authors declare no conflict of interest.



