



Autoimmune liver diseases (AILD) are a group of immunologically induced hepatic disorders that can lead to liver cirrhosis and end-stage liver disease. Extra-hepatic involvement and association with rheumatic diseases (such as Sjögren’s syndrome, systemic sclerosis and rheumatoid arthritis) are well known, whereas the coexistence of AILD with small-vessel vasculitis in the same patients have been only occasionally reported. In the present paper we report four such cases and an extensive review of the literature. Clinical features of autoimmune-liver diseases associated with small-vessel vasculitis are discussed, as well as possible common pathogenic pathways including HLA genomics, costimulatory molecules and autoantibodies. In conclusion, knowledge about this association can help physicians in recognising and treating an aggressive disease which could otherwise result in severe and multiple organ damage, compromising the overall prognosis and the indication to liver transplantation.
Autoimmune liver disorders (AILD) are a group of immunologically induced hepatic disorders including autoimmune hepatitis (AIH), primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC).1 Though hepatobiliary involvement represents the main feature of AILD, association with rheumatic diseases such as Sjögren’s syndrome, systemic sclerosis, rheumatoid arthritis has been frequently reported.2–3
On the contrary, the association of small-vessel vasculitis and AILD has been seldom reported.
We report four cases of this rare association. We also report an extensive review of the literature about the topic.
Case ReportCase 1A 75-year-old woman was admitted to the hospital after a bilateral mastectomy for tumor-like lesions. The histopathological examination of the surgical sample had shown granulomatous lesions with focal components of granulocyte-dominated vasculitis and microabscesses. Her past medical history included chronic sinusitis, nasal polyps and dacryocystitis.
The patient complained of difficulty in nasal breathing due to crusty secretions, xerostomia and xerophthalmia. The general physical examination was unremarkable.
Laboratory investigation showed mild eosinophilia (908/μL) and moderately elevated alkaline phosphatase (ALP - 1.4x) and gamma-glutaryl transferase (γ-GT - 2.5x), increased IgM (410 mg/dL) and slightly elevated total cholesterol (241 mg/dL). Kidney function tests, urine analysis and inflammatory markers were normal. The sputum culture was positive for Staph aureus.
Anti-mitochondrial (AMA) and anti-SS-A autoantibodies were present, while antinuclear antibodies (ANA) and cryoglobulins were absent. Antineutrophil cytoplasmic antibodies (ANCA) tests, performed both by indirect immunofluorescence (IIF) on ethanol-fixed neutrophils and ELISA assay for antibodies to myeloperoxidase (MPO) and proteinase-3 (PR3) were negative as well.
Ultrasound examination of the liver showed no biliary obstruction.
A percutaneous needle liver biopsy showed a moderately active chronic hepatitis with intralobular and portal granulomas, consistent with PBC (stage 3).
Nasal endoscopy revealed the presence of crusty lesions and the biopsy showed lesions similar to the previous post-mastectomy breast tissue biopsy suggestive of granulomatosis with polyangiitis (GPA, formerly known as Wegener granulomatosis).
High resolution computed tomography (HRCT) of the chest showed several nodules located around the bronchial vessels and in the subpleural space in both lung fields, which were suggestive of vasculitic lesions. There were also some small pulmonary nodules located in the upper lung fields due to post-TB scarring processes. The Mantoux test was positive but no acid fast bacilli were found in the sputum culture.
The salivary gland scintigram, the Schirmer test and the Break Up Time test were all compatible with Sicca syndrome.4
Absence of renal involvement and inflammation markers on one hand and coexisting pathologies on the other (severe osteoporosis and post-TB scarring processes) suggested a non-aggressive therapeutic management of the patient. The usual therapeutic regimen consisting in the combination of cyclophospamide with methylprednisolone was therefore excluded. Instead, we prescribed antibiotics. This decision was based on clinical evidence that has shown that nasal colonization with Staph aureus is an independent risk factor for relapse of GPA. Furthermore, prophylactic treatment with cotrimoxazole can reduce the incidence of disease relapses and has proven effective for the induction of remission in early or limited forms of GPA.
Consistently with these observations, after two weeks of antibiotic treatment (cotrimoxazole 800 mg/160 mg one tablet twice daily) our patient was asymptomatic. A HRCT of the chest performed two months later showed complete resolution of the vasculitic lesions.
Currently, three years after the diagnosis of GPA, no vasculitic relapses have occurred.
Case 2A 27-year-old man was admitted to the hospital complaining of upper abdominal pain. His past medical history was positive for type 1 AIH (diagnosed at the age of 16), chronic sinusitis, non-atopic asthma (skin prick tests negative), hypereosinophilia.
Diagnosis of AIH had been made on the basis of marked elevation of aminotransferases, hypergammaglobulinemia, positivity for ANA and anti-smooth muscle antibodies (SMA), liver biopsy showing piecemeal necrosis with plasmacellular infiltrate. He was being treated with azathioprine (50 mg/day) and low-dose methylprednisolone (4 mg/day).
On admission he was febrile; abdominal tenderness was present in the epigastric and right hypochondriac regions. Murphy’s sign was positive.
He had hypereosinophilia (1,518/μL) with normal levels of total serum IgE. Aspartate aminotransferase (AST), alanine aminotrasferase (ALT) and cholestatic liver enzymes were elevated (AST 1.2x, ALT 1.5x, gGT 3.4x, alp 1.3x). C-reactive protein (CRP) levels were also increased (5x).
ANA and SMA were positive at low titre. ANCA tests, performed both by IIF on ethanol-fixed neutrophils and ELISA assay for antibodies to MPO and PR3, were negative.
Ultrasound examination of abdomen revealed thickening of the gallbladder wall (6 mm) without evidence of gallstones.
Following the diagnosis of acute cholecystitis the patient was proposed for cholecystectomy and an open liver biopsy to evaluate the progression of AIH. Histopathological examination of the gallbladder wall revealed a marked inflammatory cell infiltrate (mainly eosinophils). No gallstone were found in the gallbladder.
Liver biopsy specimen showed chronic hepatitis of minimal activity with mild portal fibrosis and eosinophilic portal vasculitis, the latter being absent in the previous biopsy performed 11 years before.
Our patient was diagnosed as suffering from Churg-Strauss syndrome (CSS) and he was treated with azathioprine (50 mg/day) and high-dose methylprednisolone (40 mg/day). A month after his discharge he was well and without symptoms of asthma, his eosinophil count was normal.
Case 3A 72-year-old man was admitted to the hospital to investigate a previous diagnosis of ANCA-positive vasculitis established two months earlier on the basis of fever, livedo reticularis, Raynaud phenomenon, positivity for ANCA (ELISA was positive for anti lactoferrin detected with a non-specified kit in a different laboratory) and a dramatic response to steroid treatment.
On admission he was asymptomatic and his physical examination was unremarkable.
Inflammatory indexes were elevated - erythrocyte sedimentation rate (ESR) 76 mm/h, CRP 3x - and laboratory tests were compatible with cholestasis (γGT 6x, ALP 1.2x). ALT, AST and bilirubin were normal. His medication at the time included prednisone 5 mg/day.
Autoantibody profile showed medium titre positivity for ANA with a multiple nuclear dots pattern (MND); AMA were negative.
We performed an immunoblot assay to characterize liver-specific antibodies (EUROLINE assay, EUROIMMUN, Germany) which showed a specificity to Sp1OO.
ANCA tests, performed both by IIF on ethanolfixed neutrophils and ELISA assay for antibodies to MPO and PR3, were negative.
Ultrasound examination of the abdomen was normal. The patient refused to undergo liver biopsy.
In the absence of any diagnostic criteria, the hypothesis of vasculitis was discarded and steroid treatment suspended. Instead, intrahepatic cholestasis and ANA-MND positivity with Sp1OO specificity led us to a probable diagnosis of PBC (even without histologic support) and treatment with UDCA treatment was prescribed.
Six months after steroid withdrawal the patient developed acute kidney failure with microhematuria and raised inflammatory indexes (ESR 1O9 mm/h, CRP 9x, creatinine 2.78 mg/dL).
At that time ANCA were detected at a high titre both at IIF (with a peripheral pattern – pANCA) and at ELISA test (MPO+/PR3-).
A kidney biopsy was attempted but the sample was inadequate. A diagnosis of probable microscopic polyangiitis (MPA) was made and steroid treatment was prescribed (prednisone 25 mg twice daily).
Four weeks later, shortly after prednisone was being tapered on the basis of a good clinical and biochemical response, the patient complained of fever (up to 39.5 °C), cough and chest pain.
On admission he was febrile and dyspnoeic; laboratory tests showed WBC 10,770/μL, ESR 7O mm/h, PCR 3Ox, creatinine 2.58 mg/dL. HRCT detected multiple ground-glass areas located in both pulmonary fields.
The patient was treated with antibiotics and high-dose steroid therapy (methylprednisolone 6O mg/day). Nonetheless, clinical conditions worsened progressively and displacement to Intensive Care Unit was necessary. Despite aggressive immunosuppressive therapy (methylprednisolone 1 g/day for 9 days) and invasive ventilation the patient died two weeks later for respiratory failure.
Post-mortem examination revealed diffuse pulmonary consolidation due to inhalation pneumonia as the cause of death and confirmed the diagnosis of PBC and MPA.
Case 4A 36-year-old woman came to our attention as outpatient in order to investigate two episodes of angioedema.
Her past history included PBC diagnosed one year earlier on the basis of chronic cholestasis (AST 1.3x, ALT 1.2x, ALP 1.8x, γGT 11x), positivity of AMA and liver biopsy consistent with a stage 1 PBC.
Laboratory tests at that time were unremarkable, in particular peripheral white blood cell count was normal and the autoantibody-profile (including ANA and ANCA) was negative.
She was treated with H1 histamine-antagonists for the angioedema episodes and she was well for 4-5 months, after which she developed wrist and ankle arthritis, persistent rhinitis with CT evidence of sphenoid sinus hypertrophy, peripheral eosinophilia (4,OOO/mmc). The patient did not refer the appearance of this new clinical and laboratoristic abnormalities.
After one year she was admitted to hospital for anasarca with conspicuous pleural and abdominal effusion. Physical examination also revealed diffuse skin papulae.
Laboratory tests at admission confirmed marked hypereosinophilia (3,6OO/mmc), associated with increased IgE levels (348 KU/l). ALT and AST were slightly increased (1.2 and 1.3 x respectively). AMA were positive at low titre, ANA were negative.
ANCA tests, performed both by IIF on ethanolfixed neutrophils and ELISA assay for antibodies to MPO and PR3, were negative.
Echocardiogram showed Ebstein abnormality with severe tricuspid valve incompetence and moderate tricuspid valve stenosis.
A cardiac catheterism with right ventricular biopsy was performed, hystologic findings included thick and deep layers of loosely arranged collagen tissue with abundant eosinophil infiltrate.
Skin lesions biopsy was consistent with leukocytoclastic vasculitis.
The patient was diagnosed as probable Loeffler syndrome and treated with prednisone 1 mg/kg/day. Treatment dose was rapidly decreased and subsequently withdrawn due to psychiatric side effects, however the patient remained asymptomatic and without blood cell count abnormalities for over one year.
Two years later she was readmitted to the hospital for fever, dyspnoea and cough. Once again laboratory tests demonstrated hypereosinophilia (2,670/ mmc), negativity of ANA and ANCA.
HRCT detected multiple ground glass areas in both pulmonary fields. Bronchoalveaolar lavage (BAL) showed eosinophilia (70,000/mmc). BAL culture, aspergillus skin prick-test and aspergillus-specific IgE and IgG antibodies, were negative.
A diagnosis of CSS was made and the patient was treated with cyclophosphamide 50 mg/day and methylprednisololone 16 mg/day. Fever and respiratory symptoms resolved in a few days without recurrence of steroid-related psychosis and four months later HRCT of the lung documented resolution of the multiple ground-glass areas.
DiscussionCases 1, 3 and 4 meet the criteria for diagnosis of PBC according to the most recent guidelines.5 In case 2 AIH was defined by a very high score according to the International Autoimmune Hepatitis Group.6
Besides, according to the Chapel-Hill and ACR criteria for systemic vasculitis, case 1 can be classified as GPA, case 2 and case 4 as CSS, and case 3 as MPA.7–8
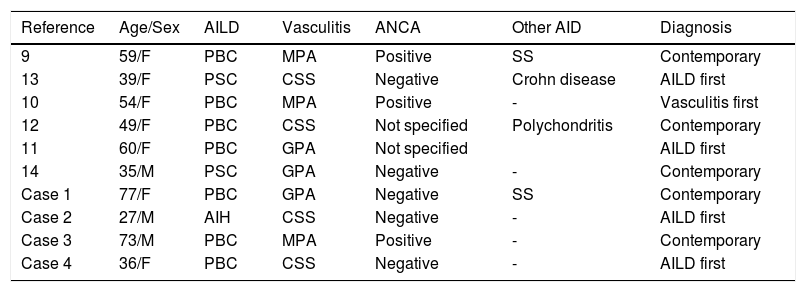
The association of small-vessel vasculitis and AILD is seldom described. As seen in table 1, our search of the literature found six other cases of this association: two cases of PBC and MPO,9,10 one case of PBC and GPA,11 one case of PBC and CSS,12 one case of PSC and CSS,13 one case of PSC and GPA.14
New and previously reported cases of association between autoimmune liver diseases (AILD) and small-vessel vasculitis.
| Reference | Age/Sex | AILD | Vasculitis | ANCA | Other AID | Diagnosis |
|---|---|---|---|---|---|---|
| 9 | 59/F | PBC | MPA | Positive | SS | Contemporary |
| 13 | 39/F | PSC | CSS | Negative | Crohn disease | AILD first |
| 10 | 54/F | PBC | MPA | Positive | - | Vasculitis first |
| 12 | 49/F | PBC | CSS | Not specified | Polychondritis | Contemporary |
| 11 | 60/F | PBC | GPA | Not specified | AILD first | |
| 14 | 35/M | PSC | GPA | Negative | - | Contemporary |
| Case 1 | 77/F | PBC | GPA | Negative | SS | Contemporary |
| Case 2 | 27/M | AIH | CSS | Negative | - | AILD first |
| Case 3 | 73/M | PBC | MPA | Positive | - | Contemporary |
| Case 4 | 36/F | PBC | CSS | Negative | - | AILD first |
ANCA: anti-neutrophil cytoplasmic antibodies. AID: autoimmune diseases. PBC: primary biliary cirrhosis. PSC: primary sclerosing cholangitis. MPA: microscopic polyangiitis. SS: Sjogren syndrome. CSS: Churg-Strauss syndrome. GPA: granulomatosis with polyangiitis. AIH: autoimmune hepatitis.
As expected, the association of these autoimmune diseases is more frequent in the female gender (7 out of 10 patients). However, male can also be affected, especially when PSC is the underlying liver disorder.
Age at diagnosis ranged from 35 to 60 years in the cases previously described. However, our cases showed that clinical presentation can occur either at an earlier age or at a more advanced one.
Small-vessel vasculitis patients with a late onset often present PBC rather than AIH or PSC. Probably also this aspect is influenced by clinical features of the underlying AILD. In fact, PBC can be considered a “late-onset disease” in comparison with PSC, which tends to affect younger patients, and AIH, which can occur at any age.
Chronological presentation of diseases seems to differ from case to case: 4 patients were diagnosed with AILD first and 1 with vasculitis first. In 5 cases the two different diseases were diagnosed simultaneously.
“Syndromic” clinical presentation is also common as 4 out of 10 patients presented at the time of diagnosis a third associated autoimmune disease (2 Sjögren’s syndrome, 1 Crohn disease and 1 polychondritis).
Scarcity of reports about the association between AILD and small-vessel vasculitis may depend on atypical clinical presentation or absence of serological markers.
Atypical clinical presentation of small-vessel vasculitis, with large arteries involvement was already described by Conn,12 but other unusual features can be found in our cases. In case 1 mammary involvement was GPA first manifestation; in case 2 clinical presentation of CSS was acute acalculous cholecystitis. Both mammary involvement in GPA15 and gallbladder inflammation in CSS16 have been described in the literature; however, this kind of atypical presentation makes the diagnosis of vasculitis even more difficult.
GPA, CSS and MPA are collectively known as “ANCA-associated vasculitis”, but cases 1, 2 and 4 were ANCA-negative, as well as some other cases reported in the past.
ANCA-negativity has been reported to be more frequent in GPA patient without renal involvement (“limited GPA”) and in CSS patients with cardiac manifestations, lung involvement, or systemic vasculitis features (whereas ANCA-positive patients are more prone to renal or peripheral nervous system involvement and alveolar hemorrhage).17
Even if not required for the diagnosis,7 ANCA are still considered a serological hallmark of these diseases and their negativity may interfere with a correct diagnosis in atypical or dubious cases.
In this series of AILD cases, ANCA absence identified a subset of patients with a less aggressive, subtle, vasculitic involvement (expecially “limited GPA” and CSS without renal impairment). On the contrary, ANCA-positive patients usually had a more extensive systemic involvement at the diagnosis; furthermore, it should be noted that the only case with a fatal outcome (case 3) was amongst the ANCA-positive patients.
As regards hepatic involvement, no difference in progression to end-stage liver disease was found between ANCA-positive and ANCA-negative patients.
Our case 3 points toward the importance of surrogate serological markers such as ANA-MND in the diagnosis of PBC, as in 15% of PBC patients AMA are undetectable using routine methods resulting in a more difficult diagnosis.18
So far, the absence of a clear explanation for the coexistence of small-vessel vasculitis and AILD has made impossible to establish if such association is causal or casual.1O Some similarities between these two groups of diseases can be found, namely in HLA-related genetic predisposition and in pathogenic pathways.
Ancestral haplotype HLA A1-B8-DR3, described many years ago as a relevant genetic risk factor for PSC and AIH,19,20 has been recently reported to be associated to GPA as well.21
Furthermore, HLA DRB1*O8 prevalence is known to be higher compared to the general population both in PBC22 and in CSS patients.21
Costimulatory molecules’ role has also been investigated in AILD and small-vessel vasculitis. Cytotoxic T-lymphocytes antigen 4 (CTLA-4) is a negative costimulatory molecule involved in maintaining tolerance and avoid autoreactivity.23 Single nucleotide polymorphisms in CTLA-4 have been suggested as genetic non-HLA related risk factor in AILD.23–25
The role of CTLA-4 polymorphisms in small-vessel vasculitis is subject of many studies at the present time: both an important role of Single nucleotide polymorphism in CTLA-4 region and increased levels of this costimulatory molecule on the surface of CD4 T-lymphocytes have been found in GPA.26–27 Taken altogether, these findings suggest that CTLA-4 alterations may be a common pathogenic pathway in the development of both AILD and systemic vasculitis.
Finally, ANCA had been proposed as a possible further link9 but more recent studies revealed different target antigens in AILD and vasculitis.
In fact ANCA in systemic vasculitis are directed against cytoplasmic antigens such as myeloperoxidase (MPO) and proteinase-3 (PR3),28 while Terjung, et al. demonstrated that ANCA target in AIH and PSC is an antigen localized in the nuclear lamina, recently identified as beta-tubulin isotype 5.29
In conclusion, the association between autoimmune liver diseases and small-vessel vasculitis is rare but possible. Such knowledge can be important in clinical practice since extrahepatic manifestation in autoimmune liver disease patients should prompt investigations for an underlying vasculitis, potentially controlling an aggressive disease which could result in severe and multiple organ damage, compromising the overall prognosis and the indication to liver transplantation.
Abbreviations- •
AILD: autoimmune liver diseases.
- •
AIH: autoimmune hepatitis.
- •
ALP: alkaline phosphatase.
- •
AMA: anti-mitochondrial antibodies.
- •
ANCA: antineutrophil cytoplasmic antibodies.
- •
BAL: bronchoalveaolar lavage.
- •
CRP: C-reactive protein.
- •
ESR: erythrocyte sedimentation rate.
- •
γ-GT: gamma-glutamyl transferase.
- •
GPA: γgranulomatosis with polyangiitis.
- •
HRCT: high resolution computed tomography.
- •
IIF: indirect immunofluorescence.
- •
MPO: myeloperoxidase (MPO).
- •
PBC: primary biliary cirrhosis.
- •
PR3: proteinase-3.
- •
PSC: primary sclerosing cholangitis (PSC).
- •
SMA: anti-smooth muscle antibodies.
None to declare.
Conflict of InterestNone to declare.



