



Atherosclerosis is characterized by lipid accumulation, inflammatory response, cell death and fibrosis in the arterial wall, and is a major pathological basis for ischemic coronary heart disease (CHD), which is the leading cause of morbidity and mortality in the USA and Europe. Intervention studies with statins have shown to reduce plasma LDL cholesterol concentrations and subsequently the risk of developing CHD. However, not all the aggressive statin therapy could decrease the risk of developing CHD. Many clinical and epidemiological studies have clearly demonstrated that the HDL cholesterol is inversely associated with risk of CHD and is a critical and independent component of predicting this risk. Elucidations of HDL metabolism give rise to therapeutic targets with potential to raising plasma HDL cholesterol levels, thereby reducing the risk of developing CHD. The concept of reverse cholesterol transport is based on the hypothesis that HDL displays an cardioprotective function, which is a process involved in the removal of excess cholesterol that is accumulated in the peripheral tissues (e.g., macrophages in the aortae) by HDL, transporting it to the liver for excretion into the feces via the bile. In this review, we summarize the latest advances in the role of the lymphatic route in reverse cholesterol transport, as well as the biliary and the non-biliary pathways for removal of cholesterol from the body. These studies will greatly increase the likelihood of discovering new lipid-lowering drugs, which are more effective in the prevention and therapeutic intervention of CHD that is the major cause of human death and disability worldwide.
Atherosclerosis is characterized by lipid accumulation, inflammatory response, cell death and fibrosis in the arterial wall, which is the pathological basis for ischemic coronary heart disease (CHD) and stroke, and is the leading cause of morbidity and mortality in the USA and other industrialized nations.1 Major risk factors for atherosclerosis include high plasma levels of low-density lipoprotein (LDL) cholesterol and lipoprotein(a), as well as low plasma concentrations of high-density lipoprotein (HDL) cholesterol.2 Because elevated LDL cholesterol levels are a major causal factor for CHD and stroke and have been a primary target of therapy for more than thirty years, the potent HMG-CoA reductase inhibitors, statins have been developed to lower plasma LDL cholesterol levels and reduce the risk of adverse cardiovascular events.3 Moreover, reducing LDL cholesterol levels to below current guideline targets further inhibits atherogenesis and decreases adverse coronary events.4-6 Many clinical studies have found that statins can reduce new adverse cardiovascular events and CHD mortality by ~ 35%, but even aggressive statin therapy can not completely eliminate cardiovascular risk. Approximately 65% of the patients treated with statins still develop adverse cardiovascular events. Therefore, additional therapeutic interventions beyond statins are strongly needed to further reduce the risk of developing CHD and stroke.7
Cholesterol is essential for all cells in the body and it is used extensively as a major structural component of cell membranes and as a substrate for the synthesis of other steroids such as bile acids, vitamin D, and sex hormones such as estradiol, progesterone, androsterone and testosterone, as well as adrenocortical hormones such as aldos-terone and cortisone. The liver and small intestine are two crucial organs for cholesterol homeostasis. Indeed, high cholesterol biosynthesis in the liver leads to more very-low-density lipoprotein (VLDL) secreted into plasma, thereby increasing plasma total and LDL cholesterol concentrations. Increased quantities of dietary cholesterol also cause plasma cholesterol concentrations to rise in most individuals. Accumulated evidence has clearly demonstrated that elevated total and LDL cholesterol levels in plasma are an important risk factor for the development of cardiovascular diseases in humans and laboratory animals.8
Because CHD is still a leading cause of death and disability in the USA and Europe, the National Cholesterol Education Program Adult Treatment Panel III guidelines9 along with the 2012 update and the American Heart Association/American College of Cardiology recommenda-tions4,5,10,11 have suggested a much lower target for plasma LDL cholesterol concentrations (i.e., < 100 mg/dL) for individuals at high risk for adverse cardiovascular events. In this way, the total number of patients requiring more aggressive cholesterol-lowering treatment increases substantially. Because the cholesterol carried in LDL particles is derived mainly from both de novo synthesis and absorption from the diet, a better understanding of the regulatory mechanisms of hepatic cholesterol biosynthesis and intestinal cholesterol absorption should lead to novel approaches to the treatment and the prevention of CHD and stroke. Therefore, despite major advances in the treatment of atherogenic lipoproteins, substantial residual risk in patients with CHD and stroke is under intensive investigation.
Many epidemiological investigations and clinical studies have clearly demonstrated that the cholesterol contained within HDL is inversely associated with risk of CHD and is a critical component of predicting its risk.12 The HDL particles were first found in the 1960s after isolation by ultracentrifugation. After a method to precipitate apoB-containing lipoproteins was established, it could determine the cholesterol content of HDL in individual healthy subjects and patients with CHD. As a result, large-scale epidemiological studies on the relationship between the plasma concentrations of HDL cholesterol and the prevalence of CHD were extensively performed. The Framingham Heart Study showed the first compelling evidence of the strong inverse association between HDL cholesterol concentrations and CHD. Based on these epi-demiological findings, a widely acknowledged concept was proposed that HDL might have properties that protect against CHD, leading to the idea that HDL is the “good” cholesterol, as opposed to LDL “bad” cholesterol. As a result, a new concept was addressed that therapeutic intervention to raise plasma HDL cholesterol concentrations would reduce risk of CHD, which was supported by a series of animal studies in the 1980s and 1990s. Subsequently, many advances were made in understanding the molecular and genetic regulation of plasma HDL metabolism.
Animal studies have found that the infusion of HDL into rabbits reduces a risk of developing diet-induced atherosclerosis.13 In addition, atherosclerosis is protected in mice overexpressing apolipoprotein A-I (apoA-I), the major HDL protein, even a high-cholesterol and high-fat diet is fed. A further study that is performed in mice with pre-existing atherosclerosis finds that overexpression of apoA-I leads to regression of pre-existing atherosclerotic disease. Taken together, these animal studies and preclini-cal results match the epidemiological investigations and clinical studies, as well as strongly support the hypothesis that HDL is a key target for a novel therapeutic approach to reducing risks of developing atherosclerosis. However, human genetic analysis and some failed clinical trials have created skepticism about the importance of HDL on the prevention and the treatment of CHD. Despite the properties of HDL consistent with atheroprotection, the causal relationship between HDL and CHD is still unclear, so far. Nevertheless, drugs that raise HDL cholesterol concentrations and other approaches that promote HDL function such as reverse cholesterol transport are being extensively investigated. Based on these findings, many new concepts and mechanisms regarding the physiological functions of HDL have been proposed.
In this chapter, we summarize recent advances in the critical role of HDL in reverse cholesterol transport and atheroprotective mechanisms, as well as in the therapeutic options of hypercholesterolemia.
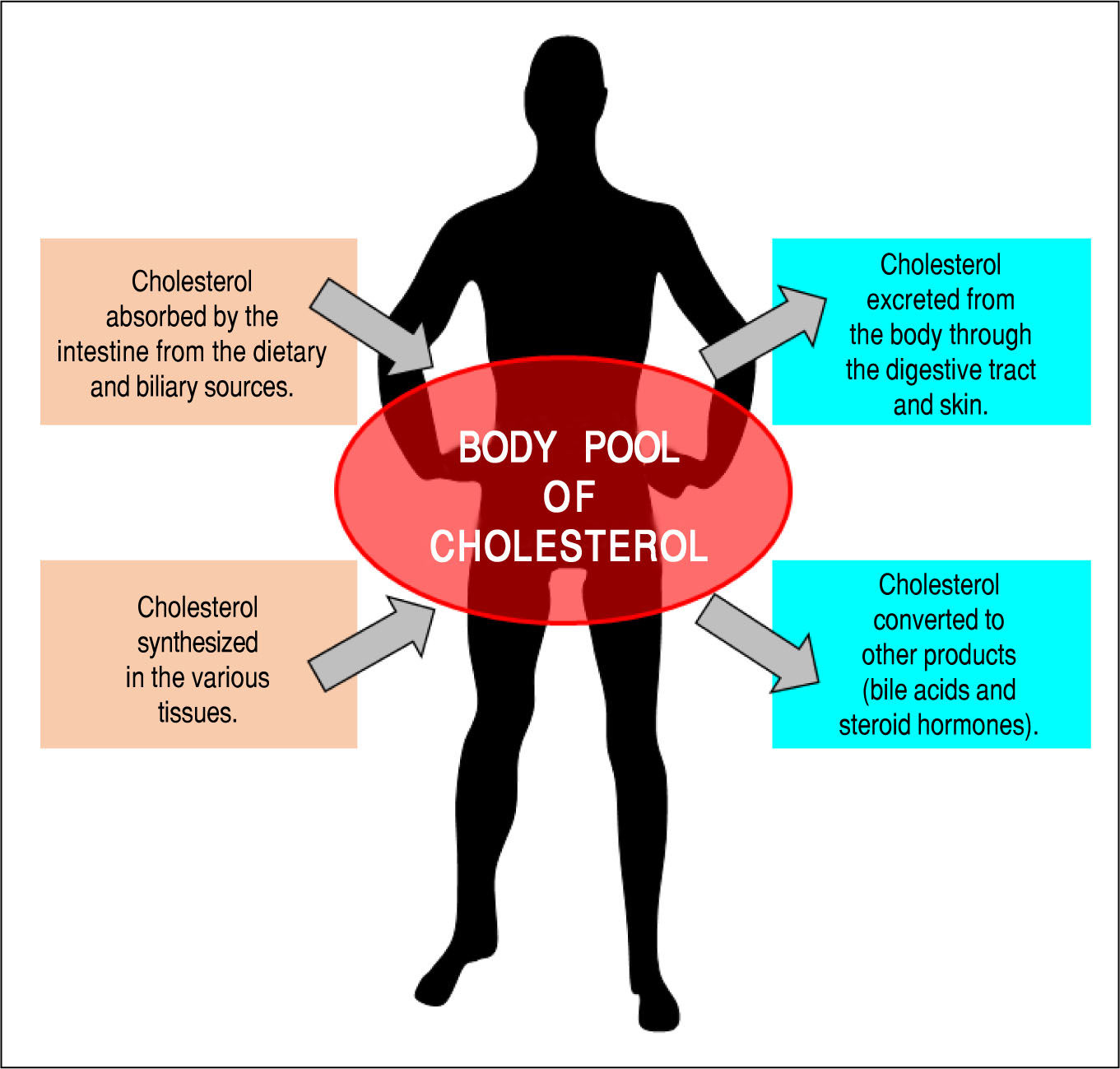
Overview of Cholesterol and Lipoprotein Metabolism in the BodyIt is well known that cholesterol is an important lipid component of virtually all cell membranes, as well as is the precursor of various steroid hormones such as the sex hormones (estrogen, testosterone, and progesterone) and corticosteroids (corticosterone, cortisol, cortisone, and aldosterone).14-17 In addition, cholesterol is largely converted into bile acids during the biosynthesis of bile acids; this step, together with the simultaneous biliary secretion of cholesterol and bile acids into bile, reduces plasma cholesterol concentrations and helps to remove excess amounts of cholesterol from the body. Figure 1 illustrates the general features of cholesterol balance across the body. Because almost all the cells in the body need a continuous supply of cholesterol, a complex series of transport, biosynthetic, and regulatory mechanisms have evolved in the human.18,19 Furthermore, cholesterol is usually obtained from the intestinal absorption of dietary and biliary cholesterol and the newly synthesized de novo from acetyl CoA within the body. However, cholesterol cannot be metabolized to CO2 and water in the body because human tissues do not possess enzymes that are able to degrade the ring structure of this sterol. Thus, to prevent a potentially hazardous accumulation of cholesterol in the human body, excess cholesterol has to be metabolized to other compounds and/or excreted in the feces. This challenging task is often done by modifying certain substituent groups on the hydrocarbon tail or on the ring structure of the cholesterol molecule. Consequently, cholesterol is largely excreted from the body either as the unaltered molecule (i.e., in both unesterified and esterified forms) or after biochemical modification to other sterol products such as bile acids and steroid hormones.
 intestinal absorption of dietary and biliary cholesterol; and (ii) cholesterol biosynthesis in the various tissues, predominantly in the liver and intestine. Likewise, there are two main pathways for the excretion of cholesterol from the body: (i) the excretion of cholesterol from the body through the gastrointestinal tract and skin; and (ii) the conversion of cholesterol to other compounds such s bile acids and steroid hormones. Because total input of cholesterol into the body must equal total output in the steady state, the body pool of cholesterol can be fundamentally kept constant. As a result, it prevents a potential accumulation of excess cholesterol in the body. Of note is that in children, there is necessarily a greater input of cholesterol into the body than output since there is a net accumulation of cholesterol for keeping body weight growth. Reproduced with slightly modifications and permission from Wang DQ-H, Neuschwander-Tetri BA, Portincasa P (Eds). The Biliary System. Morgan & Claypool Life Sciences. The 2nd. Ed. Princeton, New Jersey: 2017.")
The general feature of cholesterol balance across the body. There are two major sources of cholesterol available for the body: (i) intestinal absorption of dietary and biliary cholesterol; and (ii) cholesterol biosynthesis in the various tissues, predominantly in the liver and intestine. Likewise, there are two main pathways for the excretion of cholesterol from the body: (i) the excretion of cholesterol from the body through the gastrointestinal tract and skin; and (ii) the conversion of cholesterol to other compounds such s bile acids and steroid hormones. Because total input of cholesterol into the body must equal total output in the steady state, the body pool of cholesterol can be fundamentally kept constant. As a result, it prevents a potential accumulation of excess cholesterol in the body. Of note is that in children, there is necessarily a greater input of cholesterol into the body than output since there is a net accumulation of cholesterol for keeping body weight growth. Reproduced with slightly modifications and permission from Wang DQ-H, Neuschwander-Tetri BA, Portincasa P (Eds). The Biliary System. Morgan & Claypool Life Sciences. The 2nd. Ed. Princeton, New Jersey: 2017.
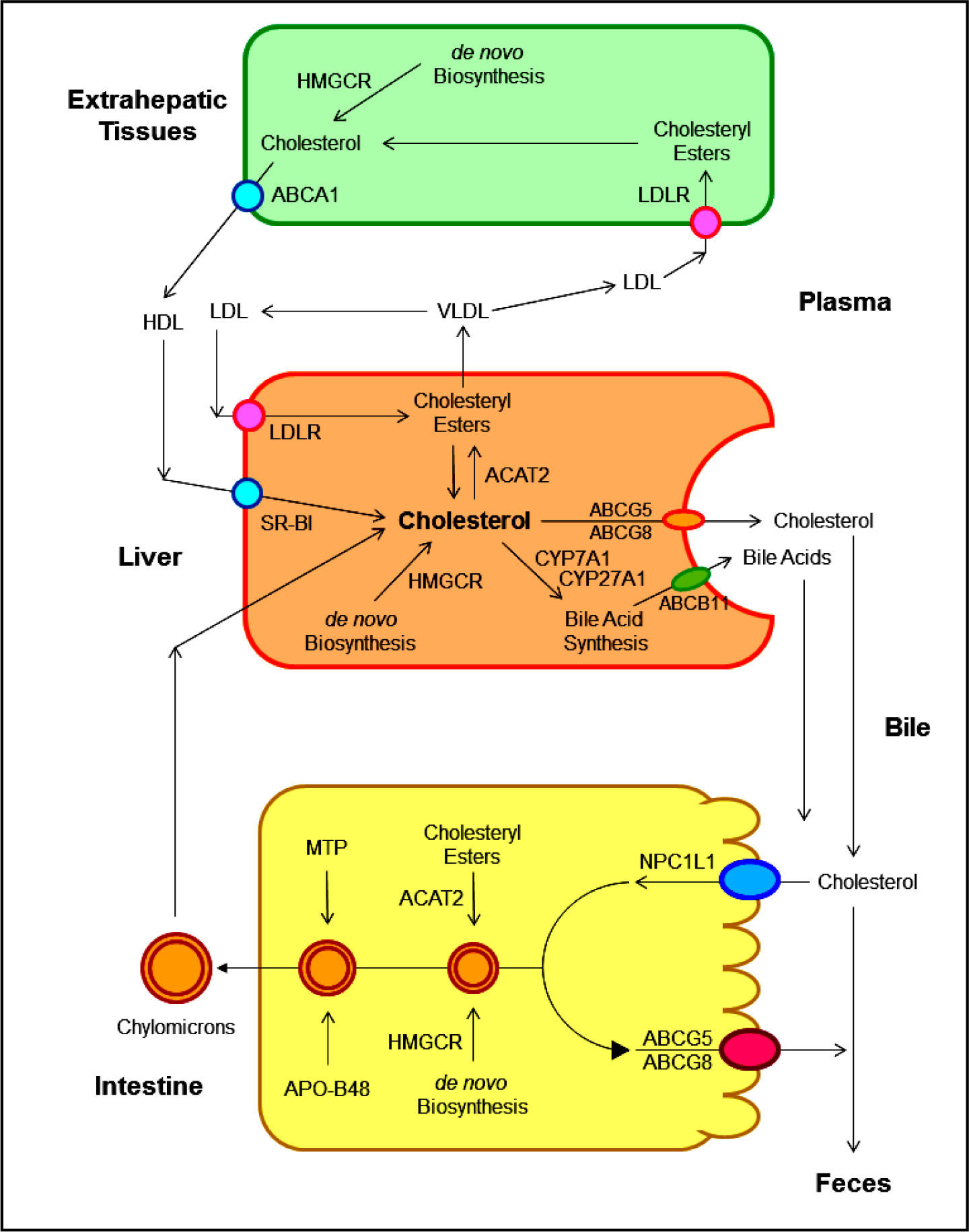
As shown in Figure 2, several pathways have been identified for the net flow of cholesterol through the major tissue compartments of the human, which illustrates how the cholesterol pool in the body is kept essentially constant in the adult.20-22 New cholesterol is added to the pool from two sources: the absorbed cholesterol from dietary and biliary sources across the epithelial cells of small intestinal tract and the newly synthesized cholesterol in a variety of different tissues within the body. The availability of dietary and biliary cholesterol to the body varies extremely in different animal species and even in the same species, and the consumed amounts of dietary cholesterol also vary strikingly from day to day.20-31 The total amount of cholesterol from the small intestine to the body also depends mainly on the absorption efficiency of intestinal cholesterol and the amount of cholesterol that is consumed daily. Furthermore, bile cholesterol is reabsorbed by the small intestine, which provides about two thirds of the total amount of cholesterol originating from the intestine every day.8 The rate of cholesterol biosynthesis in the liver varies extremely in different animal species and even in the same species. The absorbed cholesterol from the small intestine could regulate hepatic cholesterol synthesis, depending on the amount of daily food intake.
. ACAT: Acyl-coenzyme A:cholesterol acyltransferase. BA: Bile acid. C: Cholesterol. CE: Cholesteryl ester. CM: Chylomicron. LCAT: Lecithin:cholesterol acyltransferase. LDLR: Low-density lipopro-tein receptor. NPC1L1: Niemann-Pick C1-like 1 protein. Reproduced with slightly modifications and permission from Wang DQ-H, Neuschwander-Tetri BA, Portincasa P (Editors). The Biliary System. Morgan & Claypool Life Sciences. 2nd Ed. Princeton, New Jersey. 2017.")
The metabolic pathways for the net flow of cholesterol through the major tissue compartments of the human. This diagram illustrates the major pathways for the net flow of cholesterol from the endoplasmic reticulum to the plasma membrane of the cells of the extrahepatic tissues, and through the circulation to the liver where cholesterol is secreted into bile and to the intestine, and eventually, excreted in the feces. The specific transporters and receptors that are involved in these pathways are shown with arrows indicating the direction of transport. ABC: ATP-binding cassette (transporter). ACAT: Acyl-coenzyme A:cholesterol acyltransferase. BA: Bile acid. C: Cholesterol. CE: Cholesteryl ester. CM: Chylomicron. LCAT: Lecithin:cholesterol acyltransferase. LDLR: Low-density lipopro-tein receptor. NPC1L1: Niemann-Pick C1-like 1 protein. Reproduced with slightly modifications and permission from Wang DQ-H, Neuschwander-Tetri BA, Portincasa P (Editors). The Biliary System. Morgan & Claypool Life Sciences. 2nd Ed. Princeton, New Jersey. 2017.
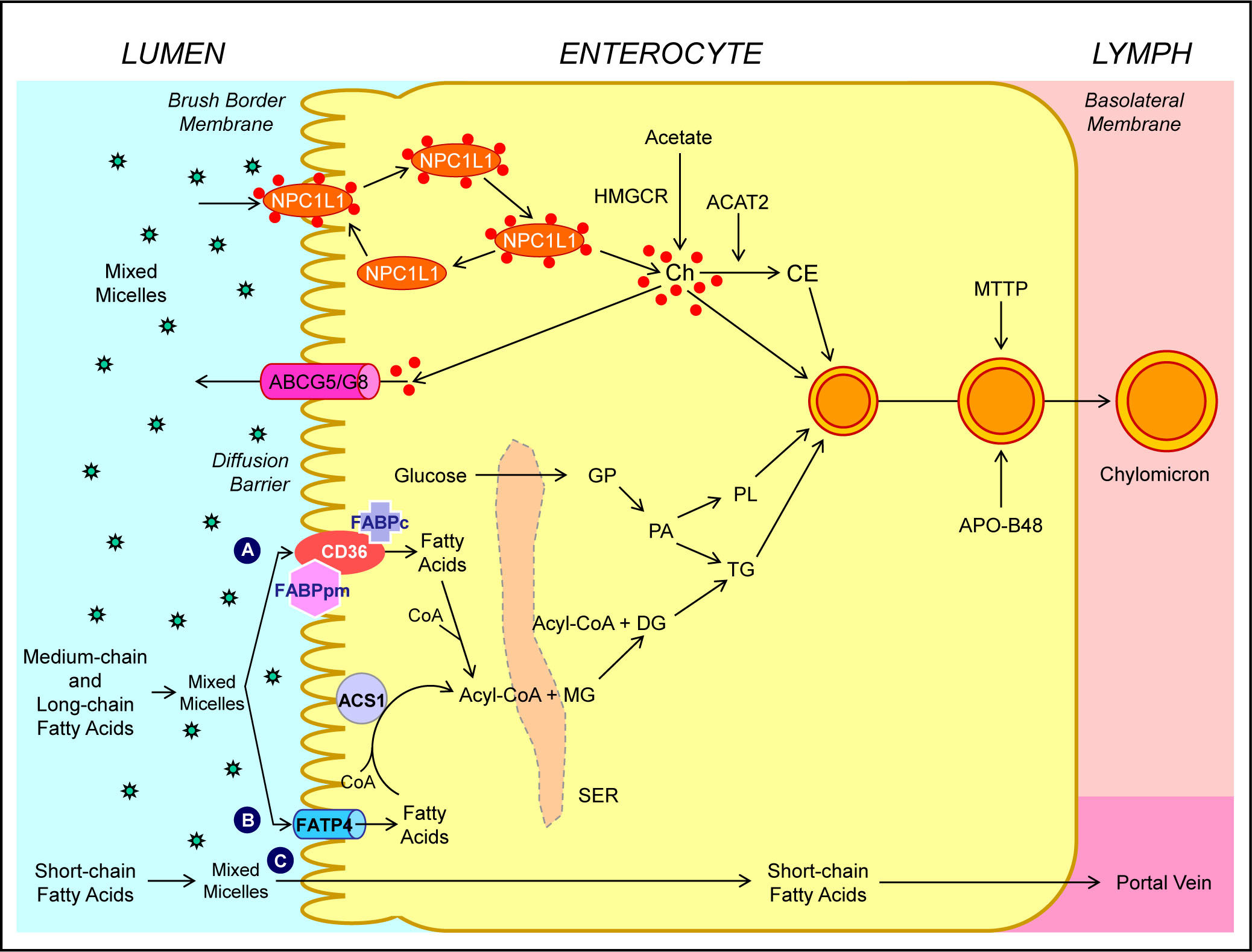
Human and animal studies have found that a transporter named the Niemann-Pick C1 like 1 (NPC1L1) protein is expressed in the apical membrane of enterocytes and plays a crucial role in the ezetimibe-sensitive cholesterol absorption pathway (Figure 3), which makes the influx of cholesterol and plant sterols from the intestinal lumen into the cytoplasm of enterocytes.32-36 In contrast, the ATP-binding cassette (ABC) transporters ABCG5 and ABCG8 are apical sterol export pumps promoting active efflux of cholesterol and plant sterols from the entero-cytes back into the intestinal lumen for fecal excretion.37-47 These findings imply that intestinal cholesterol absorption is a multistep process that is regulated by multiple genes at the enterocyte level, and that the efficiency of cholesterol absorption is determined by the net effect between influx and efflux of intraluminal cholesterol molecules crossing the brush border membrane of the en-terocyte.48 In addition, 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase is the rate-limiting enzyme for cholesterol biosynthesis in the body.49-54 Cholesterol that is synthesized de novo from acetyl CoA in different tissues (i.e., the liver and small intestine) is the second major source to the body.55-62 Therefore, the sum of these two processes constitutes the total input of cholesterol into the body pool each day.
, a sterol influx transporter, is located at the apical membrane of the enterocyte, and can actively facilitate the uptake of cholesterol by promoting the passage of cholesterol across the brush border membrane of the enterocyte. NPC1L1 appears to mediate cholesterol upt ake via vesicular endocyto-sis and ezetimibe may inhibit cholesterol absorption by suppressing the internalization of NPC1L1/cholesterol complex. By contr ast, ABCG5 and ABCG8 promote active efflux of cholesterol and plant sterols from the enterocyte back into the intestinal lumen for fecal excretion. The combined regulatory effects of NPC1L1 and ABCG5/G8 play a critical role in modulating the amount of cholesterol that reaches the lymph from the intestinal lumen. The absorbed cholesterol molecules, as well as some that are newly synthesized from acetate by 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) within the enterocyte, are es-terified to fatty acids by acyl-CoA:cholesterol acyltransferase isoform 2 (ACAT2) to form cholesteryl esters. There are three putative pathways for uptake of fatty acids and their transport across the apical membranes of enterocytes. A. Short-chain fatty acids may traverse the apical membrane by simple passive diffusion and may be absorbed into the mesenteric venous blood and then the portal vein. B. Medium and long-chain fatty acids can be transported by fatty acid transport protein 4 (FATP4). C. Alternatively, CD36 (also referred to as fatty acid translocase), alone or together with the peripheral membrane protein plasma membrane-associated fatty acid-binding protein (FABPpm; 43 kDa) accepts medium and long-chain fatty acids at the cell surface to increase their local concentrations. This could help CD36 actively transport fatty acids across the apical membrane of the enterocyte. Once at the i nner side of the membrane, these fatty acids are bound by cytoplasmic FABP (FABPc) before entering metabolic pathways. Some fatty acids may be transported by fatty acid transport proteins and rapidly activated by plasma membrane acyl-CoA synthetase 1 (ACS1) to form acyl-CoA esters. Monoacylglycerol (MG) may be taken up into enterocytes by facilitated transport. Acyl-CoA and monoacylglycerol are transported into the smooth endoplasmic reticulum (SER) where they are used for the synthesis of diacylglycerol (DG) and triacylglycerol (TG). Glucose is transported into the SER and contributes to the synthesis of phospholipids (PL) via α-glycerol phosphate (αGP). All of these lipids are involved in the assembly of chylomicrons, which also requires the synthesis of apoB48 and the activity of microsomal triglyceride transfer protein (MTTP). The core of chylomicrons secreted in lymph contains triglycerides and cholesteryl esters and their surface is a monolayer containing phospholipids (mainly phosphatidylcholine), unesterified cholesterol and apolipoproteins such asapoB-48, a poA-I and apoA-IV. Reproduced with slightly modifications and permission from Wang DQ-H, Cohen DE. Absorption and excretion of intestinal choleste rol and other sterols. In Clinical Lipidology: A Companion to Braunwald’s Heart Disease. The 2nd Edition. Editor by Ballantyne CM. Elsevier Saunders. 201 4; pp. 25-42.")
Molecular and cellular mechanisms of intestinal cholesterol and fatty acid absorption. Within the intestinal lumen, the micellar solubilization of ster-ols and fatty acids facilitates movement through the diffusion barrier overlying the surface of the absorptive cells. In the presence of bile acids, mixed micelles deliver large amounts of the cholesterol molecules to the aqueous-membrane interface so that the uptake rate is greatly increas ed. The Niemann-Pick C1 like 1 protein (NPC1L1), a sterol influx transporter, is located at the apical membrane of the enterocyte, and can actively facilitate the uptake of cholesterol by promoting the passage of cholesterol across the brush border membrane of the enterocyte. NPC1L1 appears to mediate cholesterol upt ake via vesicular endocyto-sis and ezetimibe may inhibit cholesterol absorption by suppressing the internalization of NPC1L1/cholesterol complex. By contr ast, ABCG5 and ABCG8 promote active efflux of cholesterol and plant sterols from the enterocyte back into the intestinal lumen for fecal excretion. The combined regulatory effects of NPC1L1 and ABCG5/G8 play a critical role in modulating the amount of cholesterol that reaches the lymph from the intestinal lumen. The absorbed cholesterol molecules, as well as some that are newly synthesized from acetate by 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) within the enterocyte, are es-terified to fatty acids by acyl-CoA:cholesterol acyltransferase isoform 2 (ACAT2) to form cholesteryl esters. There are three putative pathways for uptake of fatty acids and their transport across the apical membranes of enterocytes. A. Short-chain fatty acids may traverse the apical membrane by simple passive diffusion and may be absorbed into the mesenteric venous blood and then the portal vein. B. Medium and long-chain fatty acids can be transported by fatty acid transport protein 4 (FATP4). C. Alternatively, CD36 (also referred to as fatty acid translocase), alone or together with the peripheral membrane protein plasma membrane-associated fatty acid-binding protein (FABPpm; 43 kDa) accepts medium and long-chain fatty acids at the cell surface to increase their local concentrations. This could help CD36 actively transport fatty acids across the apical membrane of the enterocyte. Once at the i nner side of the membrane, these fatty acids are bound by cytoplasmic FABP (FABPc) before entering metabolic pathways. Some fatty acids may be transported by fatty acid transport proteins and rapidly activated by plasma membrane acyl-CoA synthetase 1 (ACS1) to form acyl-CoA esters. Monoacylglycerol (MG) may be taken up into enterocytes by facilitated transport. Acyl-CoA and monoacylglycerol are transported into the smooth endoplasmic reticulum (SER) where they are used for the synthesis of diacylglycerol (DG) and triacylglycerol (TG). Glucose is transported into the SER and contributes to the synthesis of phospholipids (PL) via α-glycerol phosphate (αGP). All of these lipids are involved in the assembly of chylomicrons, which also requires the synthesis of apoB48 and the activity of microsomal triglyceride transfer protein (MTTP). The core of chylomicrons secreted in lymph contains triglycerides and cholesteryl esters and their surface is a monolayer containing phospholipids (mainly phosphatidylcholine), unesterified cholesterol and apolipoproteins such asapoB-48, a poA-I and apoA-IV. Reproduced with slightly modifications and permission from Wang DQ-H, Cohen DE. Absorption and excretion of intestinal choleste rol and other sterols. In Clinical Lipidology: A Companion to Braunwald’s Heart Disease. The 2nd Edition. Editor by Ballantyne CM. Elsevier Saunders. 201 4; pp. 25-42.
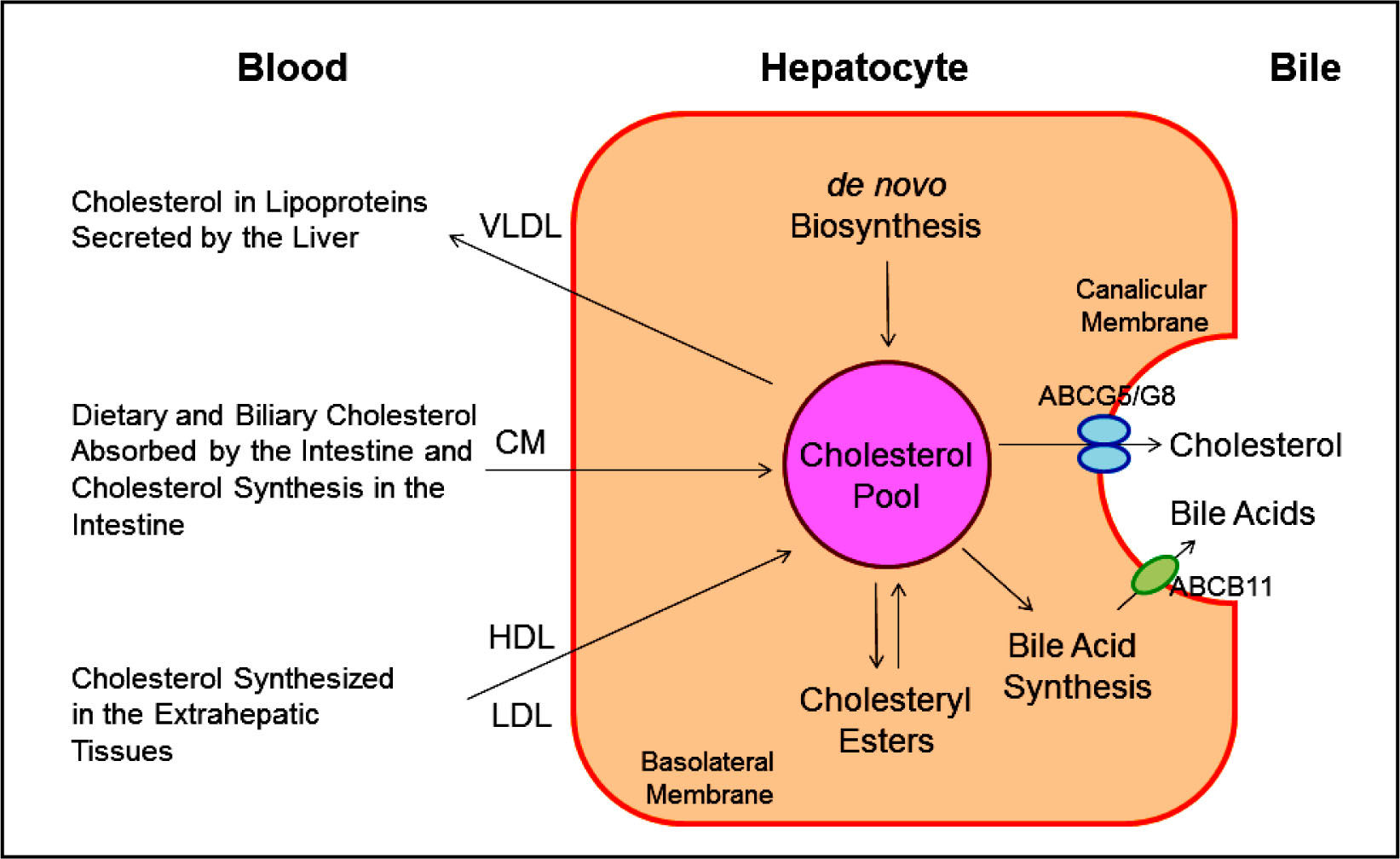
It has been found that there are two major pathways for the removal of cholesterol from the body. In humans and experimental animals, the hepatic secretion of biliary cholesterol crossing the canalicular membrane of hepatocytes is an important route for removing cholesterol from the body (Figure 4). Moreover, the cholesterol molecule can be metabolized to other compounds such as bile acids, which, in turn, are excreted from the body through the intestinal tract and eventually in the feces. Of special note is that the sterol efflux transporters ABCG5 and ABCG8 on the canalicular membrane of hepatocytesare responsible for regulating hepatic secretion of biliary cholesterol,43,63-66 and a bile acid export pump, ABCB11 is responsible for hepatic secretion of biliary bile acids.67 These transporters in the liver play a critical role in the regulation of excretion of excess cholesterol from the body, either as unes-terified cholesterol or as its metabolic products, bile acids. In addition, esterified and unesterified cholesterol is often lost directly from the body poolthrough the sloughing of oily secretions and cells from the skin, as well as through the desquamation of cells from the digestive tract.
. The Biliary System. Morgan & Claypool Life Sciences. The 2nd Ed. Princeton, New Jersey. 2017.")
This diagram shows cholesterol balance across the liver, indicating the major sources for cholesterol entering the hepatocyte and the main pathways for its disposition from the hepa-tocyte. CM: Chylomicron. Reproduced with slightly modifications and permission from Wang DQ-H, Neu-schwander-Tetri BA, Portin-casa P (Eds.). The Biliary System. Morgan & Claypool Life Sciences. The 2nd Ed. Princeton, New Jersey. 2017.
Notably, in children and growing animals, the input of cholesterol into the body is necessarily greater than the output because a net accumulation of cholesterol is vital to maintain an increased body weight. However, once adulthood is reached and body weight becomes constant, a balance between input and output of cholesterol should be kept. In other words, the input of cholesterol into the body should be equal to the output.
Taken together, the regulatory mechanisms on cholesterol metabolism must be operative, which can accurately adjust the rate of cholesterol biosynthesis within the body and the rate of cholesterol excretion from the body to accommodate the varying amounts of cholesterol that are absorbed by the small intestine at different times. Basically, these regulatory mechanisms on cholesterol metabolism work well. As a result, there is little net accumulation of excess cholesterol in the body, and yet sufficient cholesterol is always available to meet the metabolic needs of the various cells. However, delicate imbalances lead to an increase in plasma cholesterol concentration and/or hepatic cholesterol hypersecretion in humans.68-71 In the cardiovascular system, this metabolic abnormality often causes an accumulation of excess cholesteryl ester molecules within the wall of arteries, leading to clinically apparent atherosclerosis mainly in the heart and brain.72-79 In the biliary system, when an imbalance of cholesterol metabolism in bile occurs, gallbladder bile become supersaturated with cholesterol, thereby promoting the precipitation of plate-like solid cholesterol monohydrate crystals, and eventually, leading to clinically apparent cholesterol gallstone disease.80-90
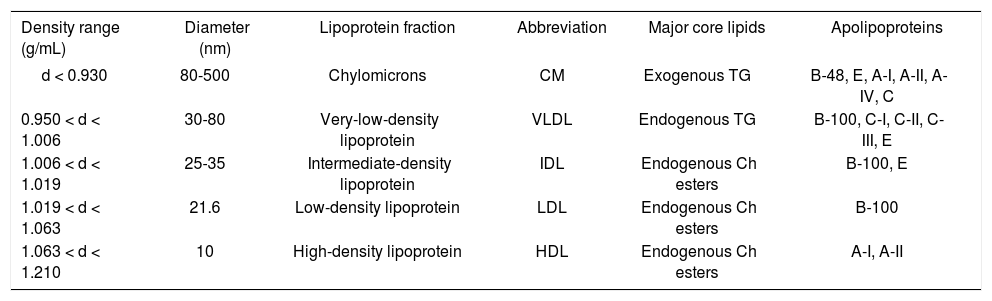
It is well known that lipoproteins in plasma include a heterogeneous mixture of particles that vary by size, density, and buoyancy (Table 1). Analytic ultracentrifugation was first applied in the 1950s to the separations of lipo-protein particles by their rates of migration in an intense centrifugal field, i.e., HDL2(F1-20 3.5-9.0), HDL3 (F1.20 0-3.5), LDL (Sf 0-12), intermediate-density lipoproteins (IDL, Sf 12-20), small VLDL (Sf 20-100), and large VLDL (Sf 100-400). These lipoprotein particles are still measured using a similar approach during its extensive uses through the 2010s. Obviously, analytic ultracentrifuga-tion is the gold standard against which other techniques are calibrated. Although the instrument is further improved to analyze plasma lipoprotein mass concentrations within individual flotation intervals, the basic methodology and the above-mentioned density intervals are still the same during their applications for basic and clinical studies.
Properties of human plasma lipoproteins.
| Density range (g/mL) | Diameter (nm) | Lipoprotein fraction | Abbreviation | Major core lipids | Apolipoproteins |
|---|---|---|---|---|---|
| d < 0.930 | 80-500 | Chylomicrons | CM | Exogenous TG | B-48, E, A-I, A-II, A-IV, C |
| 0.950 < d < 1.006 | 30-80 | Very-low-density lipoprotein | VLDL | Endogenous TG | B-100, C-I, C-II, C-III, E |
| 1.006 < d < 1.019 | 25-35 | Intermediate-density lipoprotein | IDL | Endogenous Ch esters | B-100, E |
| 1.019 < d < 1.063 | 21.6 | Low-density lipoprotein | LDL | Endogenous Ch esters | B-100 |
| 1.063 < d < 1.210 | 10 | High-density lipoprotein | HDL | Endogenous Ch esters | A-I, A-II |
In the 1950s and 1960s, a prospective study of male employees in the Livermore Radiation Laboratory was per-formed.91-94 The 10 years of follow-up found that compared to mean plasma concentrations of the total sample, the 38 men who developed CHD had 32% lower HDL2 (P < 0.01), 8% lower HDL3 (P = 0.02), 13% higher LDL (P < 0.001), 23% higher IDL (P < 0.001), and 21% higher small VLDL (P < 0.01). Plasma mass concentrations of larger VLDL were 14% greater in men with CHD than the base population.91-94 This prospective study is the first to relate HDL subfractions to the development of CHD. Moreover, it is found that the relationships of CHD to total cholesterol, to LDL, and to VLDL are reduced with increasing age, specifically after age 50.
Prospective population studies also find plasma LDL cholesterol concentrations as a positive predictor for patients with CHD and stroke.2 Furthermore, intervention studies with statins have shown to reduce LDL cholesterol levels and subsequently the risk of developing CHD and stroke.3 However, not all the aggressive statin therapy could reduce the risk of developing CHD and stroke. Further prospective population studies also identify a low HDL cholesterol level as an independent predictor for patients with CHD and stroke. This relationship persists even when LDL cholesterol levels are decreased to very low levels by therapeutic interventions with statins. Although HDL displays several potential atheroprotective properties against the development of atherosclerosis, there is still no direct evidence from clinical outcome trials in humans that raising HDL cholesterol concentrations will translate into a reduction in prevalence of CHD and stroke. Nevertheless, compelling evidence in both animal experiments and human studies demonstrate that HDL-raising therapies might reduce progression or even might promote regression of atheroma. These investigations lead to an extensive research effort to identify therapeutic interventions with the capacity to raise plasma HDL cholesterol concentrations as effectively as statins reduce plasma LDL cholesterol concentrations.
Reverse Cholesterol TransportAlthough the concept of reverse cholesterol transport was first proposed and the role of HDL in promoting this process was speculated in the mid-1960,95 the hypothesis that HDL displays an cardioprotective function was supported by numerous animal studies in the 1980s and 1990s.96-99 Many clinical studies have been performed to investigate whether raising plasma HDL cholesterol concentrations could reduce the risk of developing cardiovascular disease. Although most published reports attribute the atheroprotective properties of HDL to HDL2, many studies also find that HDL3 may be inversely related to the risk of CHD. Currently, clinical investigations of HDL focus almost exclusively on the plasma total HDL cholesterol concentrations without regard to the individual HDL subclasses. Nevertheless, elucidations of HDL metabolism give rise to therapeutic targets with potential to raising plasma HDL cholesterol levels and reduce the risk of CHD.
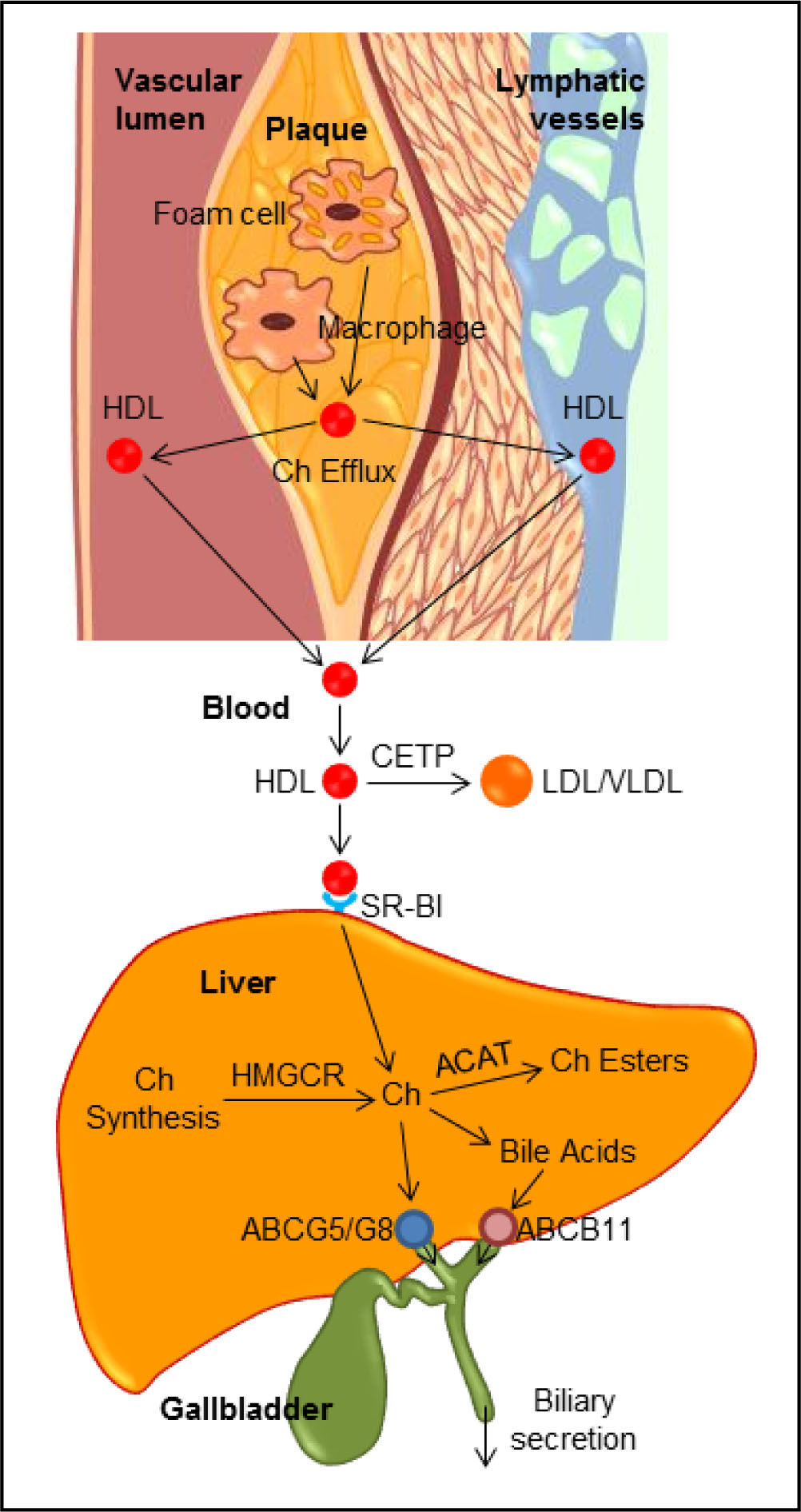
Classic Pathway of Reverse Cholesterol TransportClassically, reverse cholesterol transport is a process involved in the removal of excess cholesterol that is accumulated in the peripheral tissues (e.g., macrophages in the aortae) by HDL, transporting it to the liver for excretion into the feces via the bile (Figure 5). Plasma HDL is the smallest lipoprotein particles and contains the highest proportion of apolipoproteins to lipids compared to LDL, VLDL and chylomicrons. The major HDL-associated apoA-I and apoA-II are secreted into plasma by the liver and the intestine, where they are lipidated to form lipid poor, discoidal, nascent HDL. Nascent HDL takes up cholesterol from cell membranes and other lipoproteins. Substantial evidence from epidemiological investigations and clinical studies has clearly demonstrated that the level of plasma HDL cholesterol, especially at average to slightly above average concentrations, is inversely related to the incidence of CHD and its thrombotic complications. Prospective population studies have found that humans with HDL cholesterol levels of 6 to 7 mg/dL, i.e., higher than average, have a 20% to 27% decrease in the risk of developing CHD, and increasing HDL cholesterol levels by 1 mg/ dL may reduce the risk of CHD by 2% in men and 3% in women. Raising plasma HDL cholesterol levels has been found to retard atherogenesis and protect against atherosclerosis in mice, rabbits, and humans. Furthermore, cardiovascular risk associated with HDL cholesterol concentrations is independent of plasma LDL cholesterol levels, as well as other lipid parameters (e.g., triglycerides and total cholesterol) and non-lipid risk factors. Therefore, it was proposed many years ago that therapeutic interventions of raising plasma HDL cholesterol concentrations could potentially reduce cardiovascular mortality.100 Unexpectedly, pharmacologic interventions to increase HDL cholesterol levels by delaying HDL ca-tabolism do not translate into a marked reduction in cardiovascular risk. Thus, the inability of therapies of raising HDL cholesterol concentrations and new insights into the complexity of HDL composition and function have prompted researchers to further explore whether and how HDL exerts its cardioprotective functions.101-103

The reverse cholesterol transport through both the classic plasma pathway and the lymphatic vessel route to the liver for biliary secretion. ABC: ATP-binding cassette transporter. ACAT: Acyl-CoA:Cholesterol acyltransferase. CETP: Cholesteryl ester transfer protein. Ch: Cholesterol. HMGCR: HMG-CoA reductase. SR-BI: HDL receptor.
Although the molecular and genetic mechanisms underlying its beneficial properties in humans are not fully understood, HDL is most widely recognized for its ability to promote lipid efflux from the macrophages and other cells in the extrahepatic tissues and transport cholesterol from the periphery to the liver for biliary secretion, and subsequently, fecal excretion during the process of reverse cholesterol transport, thereby reducing the deposition of cholesterol in the peripheral tissues, including the aor-tae.104-106 Numerous animal studies have consistently found that HDL is protective on several processes that are involved in inhibiting atherosclerosis, at least in part by mediating the removal of cholesterol from lipid-laden macrophages through reverse cholesterol transport.97,107,108 When reconstituted HDL or apoA-I is provided exoge-nously, regressive changes in atherosclerotic plaques are found in human studies. Transgenic expression of the human APOAI gene increases HDL and suppresses atherosclerosis in apoE (-/) mice and genetic lowering of plasma HDL in mice reduces the appearance of macrophage-de-rived cholesterol in feces. Collectively, these results from human and animal studies have led to the idea that raising plasma HDL may be a new strategy for the treatment and the prevention of CHD and stroke.
Importance of the lymphatic pathway in reverse cholesterol transportAccumulated evidence from human and animal studies has clearly demonstrated that one of the most important biological functions for HDL is to regulate reverse cholesterol transport that promotes cholesterol transport from the peripheral tissues, including the aortae, to the liver for biliary excretion (Figure 5). This indicates that HDL plays a critical role in protecting against the development of atherosclerosis through reverse cholesterol transport. It is well known that plasma contains numerous HDL particles that are involved in reverse cholesterol transport.105 In addition, it has long been suspected that the lymphatics may also play an important role in transporting newly effluxed cholesterol from the site of efflux to the circulation because interstitial and lymphatic fluid contains a lot of HDL and apoA-I.109 It is estimated that there is a lymph transport of more than 300 mg per day in hu-mans.110 Most importantly, recent observations have found that the lymphatic vessel route is critical for efficient reverse cholesterol transport from multiple tissues, including the arterial wall.111 Removal of cholesterol by lymphatic vessels is dependent on the uptake and transcy-tosis of HDL by scavenger receptor class B type I (SR-BI) expressed on lymphatic endothelium,112 which challenges the current view that lymphatic endothelium is a passive exchange barrier for cholesterol transport. Accordingly, these findings may connect lymphatic function to atherosclerosis because lymphatic transport function may facilitate cholesterol clearance in therapies aimed at reversing atherosclerosis.113-115 However, further studies are needed to investigate whether there are differences in HDL cholesterol levels and HDL particle sizes and compositions between lymph and plasma, as well as in the regulation of the HDL-mediated reverse cholesterol transport through the lymphatic vessel route compared to the classic plasma pathway.
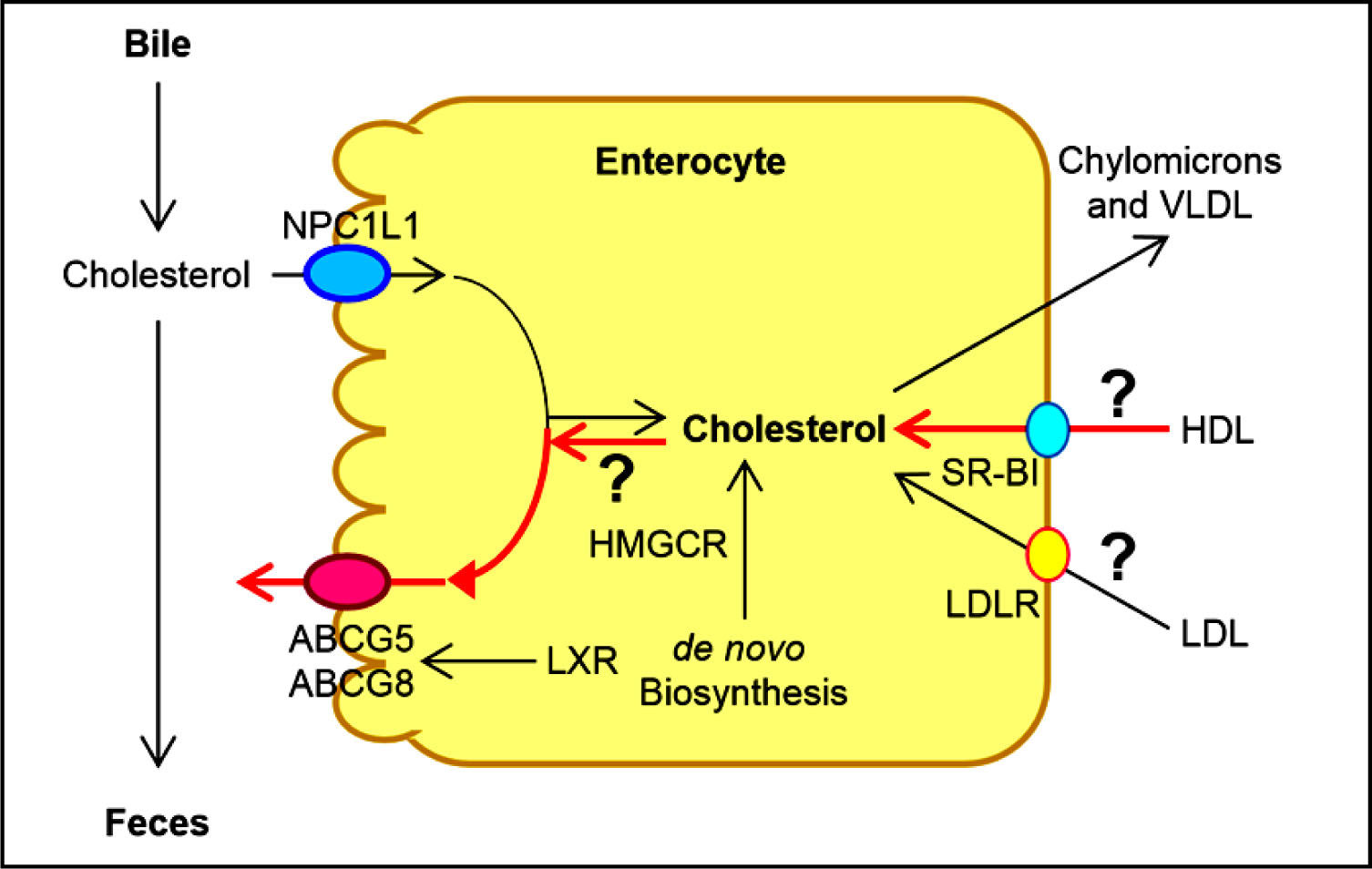
Transintestinal cholesterol excretion (TICE), a secondary, non-biliary pathway contributing to reverse cholesterol transportAs discussed above, the classic view of reverse cholesterol transport is that this important route involves the transporting of excess cholesterol accumulating within peripheral tissues back to the liver for hepatic secretion into bile, and eventually, to the intestine for excretion in the feces (Figure 6). Based on some studies on patients with hepatobiliary and/or pancreatic disorders and several animal models with obstruction of the bile duct or cholesta-sis, a novel non-biliary transport route for reverse cholesterol transport was proposedin the late 1950s.116 These studies were criticized regarding the selection of patients and animal models with the disordered condition because dramatic reduction in bile flow entering the small intestine could damage the function of the epithelial cells of intestine and lead to a striking decease in the intestinal absorption of lipids due to a lack of bile acids. Such experimental results were also questioned because these studies were performed under conditions of severe hepatobiliary disease and inappropriate experimental approaches, fecal neutral sterols in the feces are markedly increased. As a result, the transintestinal cholesterol excretion (TICE) was not accepted even though this new concept challenged the classic view of reverse cholesterol transport by showing that the intestine also significantly contributes to mass fecal neutral sterol excretion, independent of the biliary cholesterol excretion route. In the mid-2000, using different mouse models with new experimental methods, some intriguing results were reported that direct transintestinal excretion of plasma-derived cholesterol might contribute to reverse cholesterol transport in mice.117,118 Based on the results from these animal experiments, it is estimated that this non-biliary route may account for ~30% of total fecal neutral sterol excretion under basal conditions and could be modulated by liver X receptor (LXR), peroxisome pro-liferator-activated receptor-delta (PPAR-δ), and farnesoid X receptor (FXR).119-121 Subsequently, compelling evidence from animal experiments suggests that this pathway might represent a novel therapeutic target to increase reverse cholesterol transport and thereby confer protection against atherosclerosis.121 Although in vitro evidence for the activity of this pathway has been reported in explants from human small intestine mounted in Ussing chambers,122 the existence and importance of the TICE route in humans have been unclearbecause of some difficult technical issues and methodology.
 pathway, as showed in red lines. Reproduced with slightly modifications and permission from Wang DQ-H, Portincasa P, Tso P. Hepatology 2017; 66: 1337-40.")
More recently, the contribution of TICE to total fecal neutral sterol excretion is investigated in humans.123 Using a cholesterol balance approach with combined stable isotopes that label cholesterol and bile acid molecules, the body cholesterol fluxes are determined in 15 male subjects with mild hypercholesterolemia. After 4 weeks of ezetimibe (10 mg/day) treatment for inhibiting the intestinal cholesterol influx transporter NPC1L1, the same studies are performed in 10 of the subjects consuming a regular meal. Under basal conditions, approximately 65% of daily fecal neutral sterol excretion is originated from biliary secretion and the remainder (~35%) is highly likely to be derived from the TICE. Furthermore, total fecal neutral sterol excretion is increased by 4-fold in ezetimibe-treated subjects possibly through the TICE. To further confirm the results observed in the human, chow-fed Abcg8 knockout and wild-type mice are treated with ezetimibe at 0 or 8 mg/kg/day for 2 weeks. Under such experimental conditions, most of the ezetimibe-induced TICE flux is likely to be determined by the intestinal ster-ol efflux transporters Abcg5/g8. These results implied that TICE does exist in humans and most of ezetimibe-in-duced TICE flux may be regulated by Abcg5/g8. Therefore, the TICE may be a novel target to promote the removal of excess cholesterol from the body in patients at risk for CHD and stroke. It is highly likely that the TICE is an alternative route to the biliary pathway of reverse cholesterol transport. Notably, more experiments are strongly needed to investigate the molecular and cellular mechanisms underlying the critical role of the TICE in the regulation of reverse cholesterol transport in the hu-man.124 Moreover, it is still unclear whether the TICE could excrete more cholesterol from the body in patients with hypercholesterolemia, and whether the TICE works together with the classic biliary pathway, or is fully independent from the latter. More studies are also needed to explore whether the TICE is related to the absorption efficiency of intestinal cholesterol because ABCG5/G8 plays a critical role in the regulation of both the TICE and intestinal cholesterol absorption. If such a hypothesis is working, it is more interesting to explore whether there is a difference between the fasting state and the fed condition for the TICE to regulate plasma cholesterol, LDL and HDL metabolism. Obviously, TICE’s importance relative to the biliary pathway for reverse cholesterol transport is still awaiting further investigations with new experimental techniques. More importantly, whether the molecular and genetic regulation of the TICE is related to the prevalence of cardiovascular disease is still an attractive question.124 Overall, the TICE might provide a novel target for a therapeutic intervention aiming at reducing the risk of developing CHD and stroke.
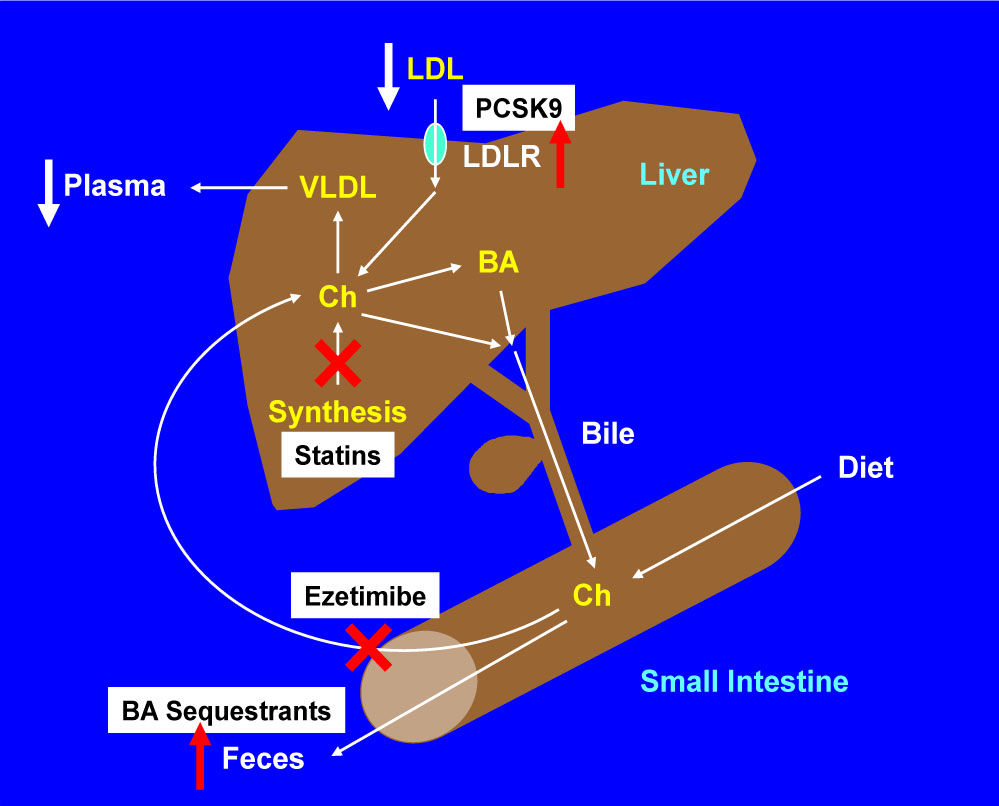
Management of HypercholesterolemiaThe potent cholesterol-lowering agents, statins have been recommended as first-line therapy in treatment guidelines of hypercholesterolemia (Figure 7), and other four classes of less commonly used compounds are often considered as second-line lipid-lowering agents, including:
- •
The intestinal cholesterol absorption inhibitors (e.g., ezetimibe);
- •
Fibrates;
- •
Niacin-based preparations; and
- •
Bile acid sequestrants (e.g., cholestyramine, resins, and colestipol).

Clinical studies have found that among these lipid-lowering agents, statins are strikingly effective in reducing risk of developing CHD and stroke, as well as in extending life when prescribed to appropriate patients.
It is well known that statins are HMG-CoA reductase inhibitors. They suppress cholesterol biosynthesis in the liver by inhibiting the enzymatic activity of HMG-CoA reductase, as well as increase the number of hepatic LDL receptors to help clear the plasma LDL (i.e., “bad” cholesterol). Available since the late 1980’s, statins, including atorvastatin, simvastatin and rosuvastatin, have been extensively prescribed to patients with hypercholesterolemia. Statins have been found to prevent cardiovascular diseases such as CHD and stroke in those subjects who are at high risk. It has clearly demonstrated that statins are effective in treating CHD and stroke in the early stages of a disease (secondary prevention). The evidence is weaker that statins are effective for those with elevated cholesterol levels but without cardiovascular diseases (primary prevention). As of 2010, four new statins are available to patients, including fluvastatin, lovastatin, pitavastatin and pravastatin, in addition to the aforementioned three statins.
The clinical benefits of statins are attributable mainly to their ability to reduce plasma levels of LDL cholesterol that is a key player driving the lipid component of athero-genesis, and that has pleiotropic effects on the pathogene-sis of atherosclerosis. However, years of clinical trials and lipid clinics have shown that not all patients respond equally or well to statin treatments, with respect to both their efficacy and adverse effects. This heterogeneity of response has prompted extensive investigation of factors that can modulate such interindividual differences, including molecular, genetic, pathological, immunological and phar-macogenomic factors. Furthermore, it is strongly desired that new lipid-lowering drugs are developed for high-risk patients.
Currently, four new classes of lipid-lowering agents are being developed through clinical trials, including:
- •
The proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, mainly monoclonal antibodies;
- •
The microsomal triglyceride transfer protein inhibitor such aslomitapide;
- •
The apoB antisense RNA agent, i.e., mipomersen; and
- •
The cholesterol ester transfer protein (CETP) inhibitors such as anacetrapib, evacetrapib, dalcetrapib and torcetrapib.
Interestingly, the development of these four new classes of agents is based on human genetic research because some rare human genetic disorders provide a crucial clue that the dysfunctional protein molecules encoded by the genes could work as the drug targets that are associated with a favorable lipid profile. As a result, the genes that encode these target proteins could serve as interesting candidates for pharmacogenomic studies of some compounds.
For example, the first case of familial CETP deficiency in Japan was reported by Inazu, et al. in the early 1990s.125 These patients have very high plasma HDL cholesterol concentrations and normal or low plasma LDL cholesterol levels, as well as they often display enhanced longevity and do not suffer from CHD. Based on these clinical observations, a new hypothesis was proposed that HDL and apoA-I may have the ability to enhances reverse cholesterol transport by promoting efflux of cholesterol from macrophages in the artery wall, inhibit vascular inflammation, and improve endothelial functions. Thus, elevating HDL and apoA-I levels has been studied as therapeutic targets for CHD. However, recent clinical trials with drugs that raise HDL cholesterol concentrations, such as CETP inhibitors and niacin, have failed to support this hypothesis.97 Therefore, HDL as a therapeutic target should be re-evaluated. Moreover, some studies find that CETP deficiency results in increased levels of dysfunctional large HDL particles that lead to premature CHD.126,127 Of note, the HDL from patients with homozygous CETP deficiency are substantially larger than normal, and these HDL particles are enriched with apoA-I, apoA-II and apoE, in contrast to very large α-1 HDL from normal individuals who contain apoA-I, but not apoA-II or apoE.128 Several in vitro experiments show that the HDL particles from patients with homozygous CETP deficiency are effective as normal HDL particles do in serving as an acceptor of both ABCA1-mediated cellular cholesterol efflux from J774 macrophages or SR-BI-mediated cholesterol efflux from Fu5AH hepatoma cells.129 Taken together, further studies are strongly needed to understand the physiology and genetic regulation underlying the major atheroprotective functions of HDL on promoting cholesterol efflux and reverse cholesterol transport.
Of note is that based on genetic studies of patients with the dysfunctional protein molecules encoded by the genes, some drugs are successfully developed. Such an example is the PCSK9 inhibitors that are a class of injectable drugs approved by FDA in 2015 because they dramatically reduce plasma LDL cholesterol concentrations by 60% when combined with a statin. The PCSK9 inhibitors are monoclonal antibodies, a type of biologic drug, which bind to and inactivate PCSK9 protein in the liver. By inactivating PCSK9 via inhibition, more LDL receptors are available to capture LDL for removal from the circulation, thereby reducing plasma LDL cholesterol concentrations.
Clinical studies have found that statins are not able to completely lower plasma cholesterol levels in approximately one fifth of patients, probably because of serious genetic defects. In addition, some patients stop their statin treatment due to severe side effects. The PCSK9 inhibitors may be used alone or in combination with statins to further reduce refractory high LDL cholesterol levels in patients who cannot tolerate any statins due to severe side effects.
Future DirectionsAlthough accumulated evidence has clearly demonstrated that statins reduce the development of CHD and stroke, other lipid-lowering therapies are often used ad-junctively when statin therapy is inadequate or as an alternative for patients who are intolerant of statins. More recently, intensive lipid and pharmaceutical studies lead to considerable development of new agents that have novel targets in the metabolic pathways of lipoproteins and that have the potential to serve as new alternative or adjunctive agents to existing drugs such as statins. Because of their early stages of drug development, and the limited numbers of patients studied and the limited duration of clinical trials, there are few pharmacogenomic data for these compounds so far. Studies in patients receiving these new classes of lipid-lowering agents, especially in individuals with monogenic disorders of lipid and/or lipoprotein metabolism, will certainly provide a great opportunity to find that genotype predicts response to lipid-lowering therapy and thus guides the choice of drug and dose for high-risk patients, and especially, for patients with the hardest-to-treat elevated cholesterol concentrations due to intolerance to any statins and severe drug side effects.
In the future, the personalized medicine will need to be developed for the prevention and the treatment of cardiovascular disease that is highly prevalent worldwide. The ideal application of lipid-lowering drugs would be to identify patients at risk for either a suboptimal response with respect to efficacy, or a marked adverse response to either a drug class or a specific drug. As a result, individuals who would be predicted to have an unfavorable benefit-to-risk ratio can be identified and might be obtained from alternative methods more expeditiously and without the trial-and-error process that typically accompanies initiation and maintenance of such commonly used treatment.
Although the pharmacogenomics of lipid-lowering drugs have advanced considerably and a few consistent trends on the therapy of cardiovascular disease have emerged, mainly relating to the genetic determinants of response to statins, many new molecular, genetic and biochemical studies on lipid and lipoprotein metabolism are being extensively explored. First, it is more interesting to investigate the mechanisms of elucidating how cholesterol is excreted from the body at a molecular level. Second, the potential molecular and genetic mechanisms underlying the removal of cholesterol from the body through the biliary pathway (i.e., the classic reverse cholesterol transport) and the non-biliary routes (i.e., the TICE) are desired to be revealed. Third, it is important to understand why the genetic determinants of pharmacokinetics are more relevant than genetic determinants of pharmacodynamics in determining interindividual variation in both efficacy and adverse effects of statins and other lipid-lowering drugs. Progress in our understanding of lipid, HDL, LDL and VLDL metabolism, as well as the biliary and the non-biliary pathways for removal of cholesterol from the body will greatly increase the likelihood of finding new lipid-lowering therapies and proving that they are more effective in the prevention and therapeutic intervention of cardiovascular disease that is the main cause of human death worldwide.
Abbreviations- •
ABC: ATP-binding cassette (transporter).
- •
Apo: apolipoprotein.
- •
CETP: cholesteryl ester transfer protein.
- •
CHD: coronary heart disease.
- •
HDL: high-density lipoprotein.
- •
HMG-CoA: 3-hydroxy-3-methylglutaryl CoA (re-ductase).
- •
LCAT: lecithin:cholesterol acyltransferase.
- •
LDL: low-density lipoprotein; NPC1L1, Niemann-Pick C1 like 1 (protein).
- •
PCSK9: proprotein convertase subtilisin/kexin type 9.
- •
SR-BI: scavenger receptor class B type I.
- •
TICE: transintestinal cholesterol excretion.
- •
VLDL: very-low-density lipoprotein.
Nothing to report.
AcknowledgmentsThis work was supported in part by research grants DK106249, DK041296 and AA103557 (to DQ-HW), DK92779, and DK95440 (to M.L.), all from the National Institutes of Health (US Public Health Service).



