



Congenital choledochal cyst is malformation of the biliary ductal system, which rarely occur. We describe here a 4-month old boy, who was referred to our center with respiratory distress and low level consciousness. In physical examination, a mass was detected in right upper quadrant of abdomen. Sonographic examination indicated a cystic structure representing the choledochal cyst. Further evaluation confirmed the diagnosis of cystic fibrosis in this patient. Although choledochal cyst is considered as a rare disease, it is the most frequent malformation of the extrahepatic biliary ducts, which easily could be misdiagnosed.
Congenital choledochal cyst is malformation of the biliary ductal system, which is rare condition with incidence of incidence of about 1 in 2 millions live birth, but the most frequent malformation of the extrahepatic biliary ducts.1-3 It is as result of the embryological arrest of the normal migration of the pancreaticobiliary junction towards the duodenal wall.2 Choledochal cysts could be associated with distal biliary atresia in neonates and infants, which typically presents with cholestatic jaundice and acholic stools characteristic of biliary obstruction.3 Meanwhile cystic lesions of the extrahepatic bile duct could be the common manifestation of biliary atresia and choledochal cyst.4 The classical triad of right upper quadrant pain, jaundice and abdominal mass is present only in a few instances.5 Choledochal cysts are often misdiagnosed; however early diagnosis and appropriate treatment lead to excellent prognosis.1
We here describe a rare condition of choledochal cyst in an infant with cystic fibrosis (CF).
Case reportThe patient is a 4 month-old boy, the first child of not-related parents, who was well until the age of 4 month, when he was referred to our referral center in Tehran (Children’s Medical Center Hospital) with respiratory distress and low level of consciousness. He had fever and tachypnea.
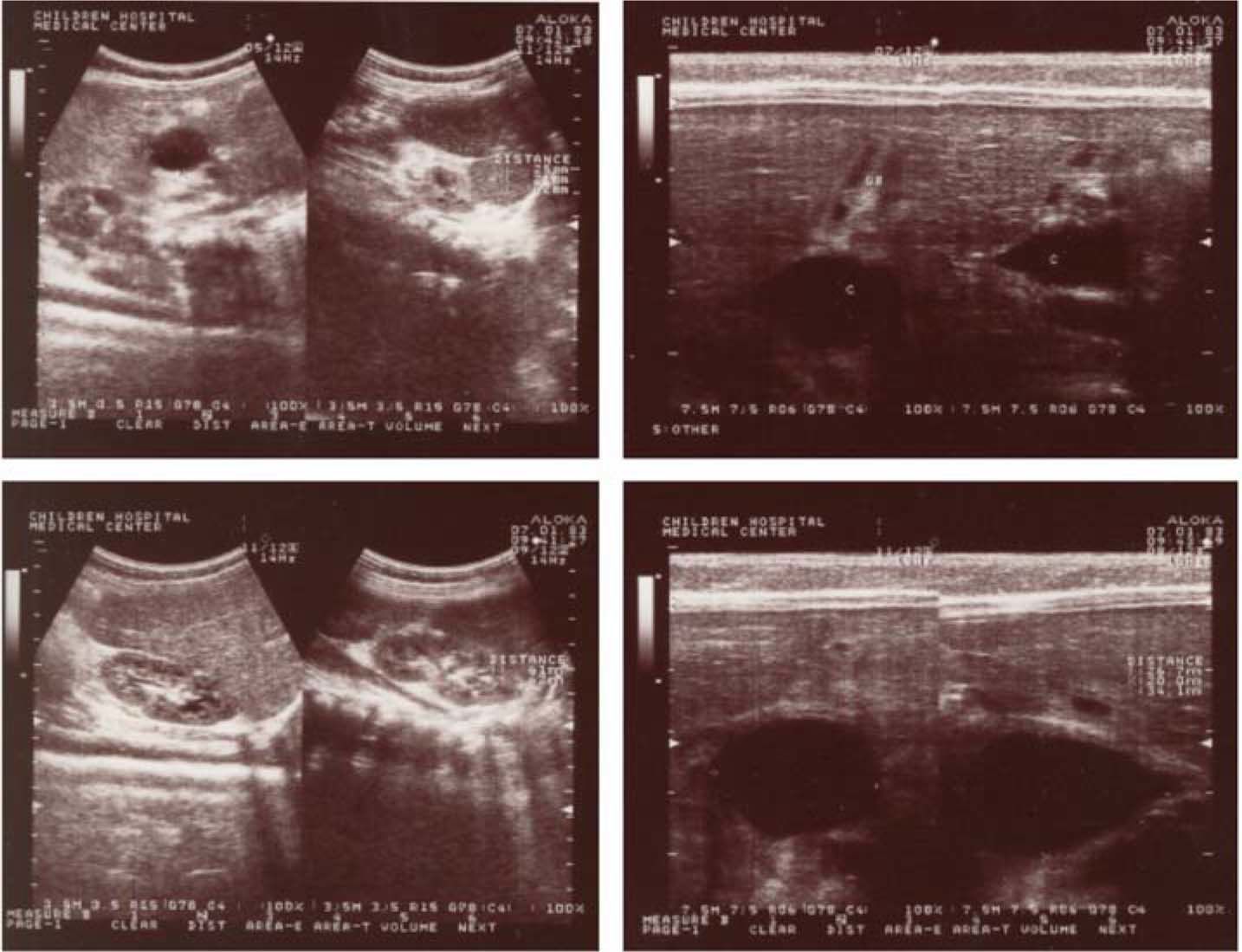
Abdominal mass was detected in right upper quadrant of abdomen, in physical examination. Laboratory studies were normal, except anemia (Hemoglobin = 7.4 g/dL). Cerebrospinal fluid (CSF) analysis and liver function test (LFT) were also normal. Chest X-ray did not show any specific finding. Abdominal sonography revealed a cystic structure indicating the choledochal cyst (Figure 1). As of suspicious to CF, sweat tests were performed twice, which confirmed the diagnosis of CF.
 near gallbladder, indicating the choledochal cyst.")
Ceftazidime (30-50 mg/kg) and Gentamicin (2 mg/kg) was administrated in addition to inhaled beta2-agonist. The patient responded to this therapy. Total excision of the extrahepatic bile duct with Roux-en-Y hepaticoje-junostomy was also performed for the patient. He was consequently discharged with good clinically condition.
DiscussionCystic fibrosis (CF) is an autosomal recessive disease of mucus and sweat glands which mainly affects the respiratory and gastrointestinal systems. The abnormal mucus could lead to a variety of clinical symptoms, including recurrent infections of airways, obstruction the duct of the pancreas, disorder of bile secretion, and the gastrointestinal obstruction. However, CF can present with different clinical features and severity from patient to patient.6 Gastrointestinal manifestation, especially hepatobiliary involvement, is one of the most important concerns in this disease.6,7 However, choledochal cyst is not expected in this disease as of a rare condition.
Congenital choledochal cysts consist of congenital dilation of the extrahepatic bile duct with a variable amount of intrahepatic involvement.2 While the clinical symptoms could be non-specific and the classical triad of disease is rarely seen5 it is often misdiagnosed.1 Although, high levels of bilirubins, serum transaminase (SGOT and SGPT), and amylase are expected in more than half of the cases,8 such laboratory studies in our case were normal.
Ultrasonography, as a rapid and accurate method for early diagnosis, is considered a very good screening diagnostic tool.2,9 Sonography could also offer the opportunity for planning the surgery before the onset of complications. Meanwhile endoscopic retrograde cholangiopancreaticography (ERCP), computer tomography (CT) and magnetic resonance cholangiopancreaticography (MRCP) have some benefits in the classification of the disease.9
The treatment of choice for choledochal cyst is surgical, which consists of the resection of the whole anomalous choledochus and definitive separation of bile ducts from the pancreatic duct.8 Complete excision of the diseased bile duct prevents development of carcinoma of the bile duct.8 Prognosis depends on early diagnosis, complete excision of the cyst, and reconstruction by hepaticojejunostomy.10 Management of our cases was successful after the complete removal.
Although choledochal cyst is considered as a rare disease, and its coincidence is very rare, it should be considered in the differential diagnosis list of unclear upper abdominal pains, jaundice and pancreatitis in children, while early diagnosis and appropriate treatment result to excellent prognosis and delay diagnosis leads to complications and even death of patients.



