




Aim. Recent studies have suggested miRNA dysregulation in liver tissue mediates the pathogenesis of various liver diseases especially liver fibrosis, but the microRNA changes during PS-induced hepatic fibrosis are still unknown. The purpose of this study was to screen the miRNA differences in rat liver fibrosis model and clarify the relationship of miRNAs with the development of PS-induced liver fibrosis.
Material and methods. Two fibrotic and two normal liver tissues from 20 Sprague-Dawley rats were collected and sequenced. MiRNA profiling results and fibrosis-related genes were validated by quantitative real-time polymerase chain reaction (qRT-PCR) and bioinformatics was used to predict miRNA targets.
Results. In total, 48 miRNAs were detected to be aberrantly expressed in fibrosis tissue compared to normal tissue. Further functional analysis of the deregulated miRNA targets revealed the miRNAs are involved in several biological functions and pathways. In addition, the expression level of miR-27a and miR-146b and fibrosis-related genes were significantly up-regulated by using qRT-PCR in fibrotic liver tissues when compared to the normal liver tissues.
Conclusion. PS-induced hepatic fibrosis results in up-regulation of the miR-27a and miR-146b in liver tissues, suggestingmiR-27a and miR-146b would be associated with the development of PS-induced liver fibrosis and be potential therapeutic targets during hepatic fibrosis.
Hepatic fibrosis is a worldwide medical problem with significant morbidity and mortality,1 and can occur as a result of viral infection, alcohol abuse, or metabolic disease.2 Activated hepatic stellate cells (HSC) play a pivotal role in the progression of liver fibrosis and is known to be mainly induced by abnormal expression of transforming growth factor(TGF)-β signaling pathway,3 which strongly induced expression of fibrosis-related genes include a matrix degrading complex comprised of collagen(COL) 1-α1, tissue inhibitors of metallopro-teinases-1(TIMP-1).4 Advanced liver fibrosis that goes no effectively treatment results in liver cirrhosis or hepatocarcinoma (HCC) and is irreversible; in contrast, hepatic fibrosis is generally reversible.5 Understanding the mechanisms of progression of liver fibrogenesis and developing effective anti-fibrotic agents are of vital importance and can prevent progression to cirrhosis and HCC.
MicroRNAs (miRNAs) represent a class of small RNAs that function as post-transcriptional regulators of gene expression.6 They play crucial regulatory roles in most cellular and developmental processes and have been implicated in many diseases in humans and animals.7,8 In the liver, aberrant expression of miRNAs are involved in different stages of the diseases.9–11 Recently, accumulating studies have demonstrated that miRNAs played an important role in the progression of liver fibrosis. miR-19b, a negative regulator for TGF-β signaling, was confirmed by decreased expression of type I collagen and by blocking TGF-β-induced expression of α1(I) and α2(I) procollagen through targeting TGF-β receptor II (TGFβRII) and smad3 expression.12 A dramatic increase in hepatic miR-199a, miR-199a*, miR-200a and miR-200b resulted significant induction of several ?brosis related genes (TIMP1, a1-pro-collagen), and was shown to be linked with the grade of liver fibrosis.4
The primary mechanism of chronic liver injury in human is closely related to immune response,13 and is obviously different from most of liver fibrosis animal models which are post-necrotic hepatic fibrosis models.14,15 The PS-induced hepatic fibrosis model is regulated by immunologic mechanisms through heterogeneous serum administration and shares several characteristics with human fibrosis of various etiologies.16,17 Thus, we employed the PS-induced fibrosis model. Recently, the miRNA expression patterns in carbon tetrachloride(CCl4) or dimethylnitro-samine (DMN)-induced rat models of liver fibrosis have been fully elucidated.14–15 However, the expression profiles of microRNAs in PS-induced hepatic fibrosis model are not yet been studied. Due to the essential roles of dysregulated miRNAs in liver physiology and disease,18–20 we hypothesized that altered miRNA expression patterns may contribute to PS-induced hepatic fibrogenesis. To test this hypothesis, we therefore examined miRNA expression profiles in PS-induced fibrotic and normal rat liver samples by deep sequencing. A further analysis of miRNA-regulated signaling pathways and related targets was performed on the basis of bioinformatics interpretation. Moreover, we also observed the key miRNAs (miR-27a and miR-146b) are associated with the development of hepatic fibrogenesis.
Material and MethodsAnimal modelsAdult rats were maintained and experimental procedures performed in compliance with the Guide for the Care and Use of Laboratory Animals of the Chinese Academy of Sciences and approved by the Medicine Animal Care Committee of Xiangya Medical College, Central South University (Changsha, HunanPR, China). Eight-week-old male Sprague-Dawley rats (120 ~ 150g; from the Department of Laboratory Animal Science, Xiangya Medical College, Central South University, Changsha, HunanPR, China) were used in this study. Twenty male rats were divided into normal control and fibrosis model groups (n = 10 per group). The fibrosis model group received twice- weekly 0.5ml intraperitoneally injections of porcine serum for 12 weeks.17,21 The control group received only saline(NS). At the end of 12 weeks, the rats were sacrificed. Liver tissues were frozen after surgical removal and stored at -80 °C.
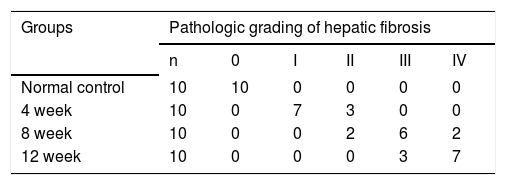
HistologyLiver tissues were fixed in 10% formaldehyde in NaCl/Pi buffer (pH 7.4), dehydrated in alcohol, and embedded in paraffin. Sections (4 μm) were prepared and stained with hematoxylin-eosin (HE) and Mas-son trichrome(MT) stain. Fibrosis was staged calculated as described:22 grade 0, normal; grade I, light fibrosis: collagen fibers expand from the limited portal area to the perisinusoidal and lobular region; grade II, mild fibrosis: mild collagen fibers extend around the portal area, form fibrous septa, but lobular structures are preserved; grade III, moderate fibrosis: collagen fibers form fibrous septa associated with lobular distortion without cirrhosis; Grade IV, severe fibrosis: early stage cirrhosis. Histopathology was independently performed by 2 board-certified pathologists who were blind to the study.
MiRNA sequencing and data statisticsTotal RNA was extracted from liver tissues with TRIZOL (Invitrogen, Carlsbad, CA, US). The concentration and quality of the purified RNAs were evaluated on a Bioanalyzer 2100 (Agilent Technologies, PaloAlto, CA). Libraries were constructed using the Illumina Small RNA Sample Prep Kit V1.0. Small RNAs ligated with adaptors were amplified by RT-PCR (15 cycles of amplification) to produce sequencing libraries. PCR products were purified on a non-denaturing acrylamide gel and sequenced for 50 cycles on the IlluminaHiSeq 2000 sequencer.
After trimming the low quality sequences and removing the adapters, the remaining reads were aligned to rat genome sequence (Rattusnorvegucus assembly, rn5) using SOAP2.23 BLAST was adopted to identify known miRNAs (miRBase 20) and other type RNAs (rRNA, tRNA, snRNA, snoRNA, GenBank&Rfam). We normalized all miRNA reads as transcripts per million clean tags (TPM) and employed the log2 (Fold-change) value with P-value ≤ 0.05 as indicator to distinguish differentially expressed miRNAs in normal and fibrotic liver tissue.24–26
GO and KEGG pathway enrichment analysisTo reduce the false positive rate, three algorithms (miRWalk, TargetScan and miRanda)27–29 have been used to identified the potential targets of deregulated miRNAs. Only the intersection of the results from these three tools was selected as representing the robust targets and used for further analysis. Gene Ontology (GO) analysis was applied in order to organize miRNA-related genes into hierarchical categories on the basis of cell component, biological process and molecular function. The target genes were annotated with GO terms using Blast2GO.30 The histogram of GO term categories was performed by WEGO.31 All putative targets were subjected to KEGG pathway annotation using DAVID Bioinfor-matics Resources (http://david.abcc.ncifcrf.gov/).32 KEGG pathways with the threshold EASE Score, a modified one-tail Fisher’s Exact Probability value ≤ 0.05 are considered strongly enriched miRNA-regulated pathways.
Quantitative Real-Time PCR (qRT-PCR) analysismiR-27a and miR-146b cDNA weresynthesised from 2ug of total RNA with an All-in-one™ miRNA First-Strand cDNA Synthesis (GeneCopoeia, Rockville, MD, USA) Kit using the supplied poly-A primer. Real-time PCR was performed in a 20ul reaction mix including 2ul of 5 x diluted reverse transcription product, 2ul miProfile miRNA qPCR primer, 2ul miRNA specific primer, 10ul SYBR 2x All-in-one qPCR Mix, 0.4ul 50 × ROX Reference dye, and 3.6ul double distilled water. The cycling conditions for amplification on the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) were 95°C for 10min, followed by 40 cycles of 95 °C for 10 sec, 60 °C for 20 sec, and 72 °C for 10 sec. The data were normalized against the U6 snRNA.
TGF-β1, TIMP1 and COL-1α1 expression were analyzed with THUNDERBIRD SYBR qPCR Mix (ToYoBo, Osaka, Japan). cDNA was synthesized with the RevertAidTM First Strand cDNA Synthesis Kit (MBI Fermentas, Ontario, Canada) in a total volume of 20μl The qRT-PCR primer sequences were designed for three fibrosis-related genes and GAPDH:
- •
For TGF-β1,5’-AGAAGTCACCCGCGTGCTA-3’ (forward) and 5’-TGTGTGATGTCTTTGGTTTTGTCAT-3’ (reverse).
- •
For TIMP1, 5’-GGGCTACCAGAGCGATCACTT3’ (forward) and 5’-AAGGTATTGCCAGGTGCACAA-3’ (reverse).
- •
For COL-1a1, 5’-GCATCAGGGTTTCAGAGCA-3’ (forward) and 5’-CGTTGGGTCATTTCCACAT-3’ (reverse).
- •
For GAPDH, 5’-CTTCCGTGTTCCTCCCC-3’ (forward) and 5’-GCATCAAAGGTGGAAGAAT-3’ (reverse).
qRT-PCR was performed on an Applied Biosystems 7500 RT-PCR System(Applied Biosystems, Foster City, CA). The cycling conditions for amplification were 95 °C for 5 min, 35 cycles of 95 °C for 30 sec, and 55 °C for 30 sec, and 72 °C for 10 sec. The data were normalized against GAPDH.
Each sample was analyzed in triplicate. Fluorescence signal was measured at each extension step. The relative expression was determined with the 2ΔΔCT method,33 where the normalized CT was calculated by subtracting the CT of a control gene (U6 snRNA for miRNA and GAPDH for mRNA) from the CT of the gene of interest.
Statistical analysesThe relationship between miR-27a, miR-146b and TGF-β1, TIMP1 and COL-1α1 mRNA expression were analyzed by Spearman’s correlation. The other data was analyzed by two-samp t-test. All data was expressed as means and standard deviation from at least 3 independent experiments. Differences with P < 0.01 (**) were considered statistically significant. The results were analyzed by SPSS17 (SPSS, Chicago, IL, USA).
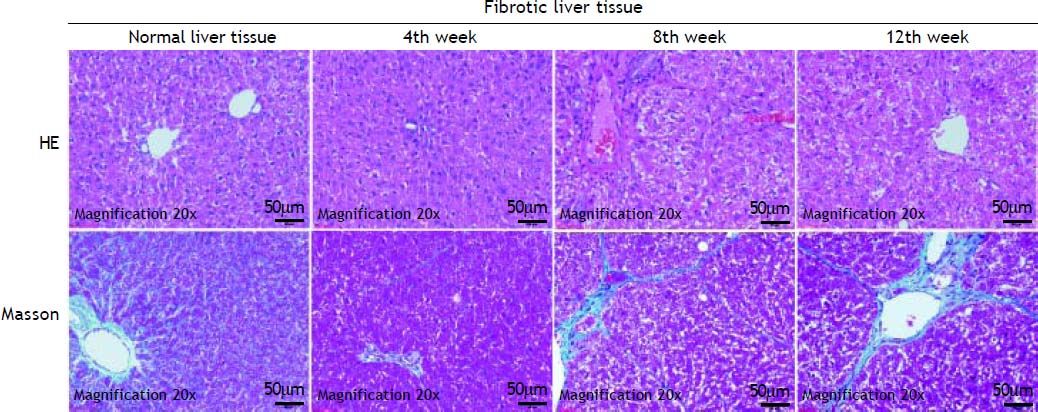
ResultsHepatic histopathology and fibrosis-related genes changes during PS- induced liver fibrosisHistopathology revealed few necrotic cells and little fibrosis in the liver of normal rats (Figure 1). In contrast, fatty degeneration, necrosis, and inflammatory cell infiltration were clear in the fibrosis model group. MT staining showed nodular fibrosis with extensive collagen deposition and well-delineated fibrotic septa, including bridging in portal regions and pseudo-lobules formation. The degree of rat hepatic fibrosis was determined by microscopy at 4, 8 and 12 weeks (Figure 1 and Table 1).
. HE staining of normal and fibrotic liver tissue at 4th week, 8th week and 12th week; Masson staining of normal and fibrotic liver tissue at 4th week, 8th week and 12th week.")
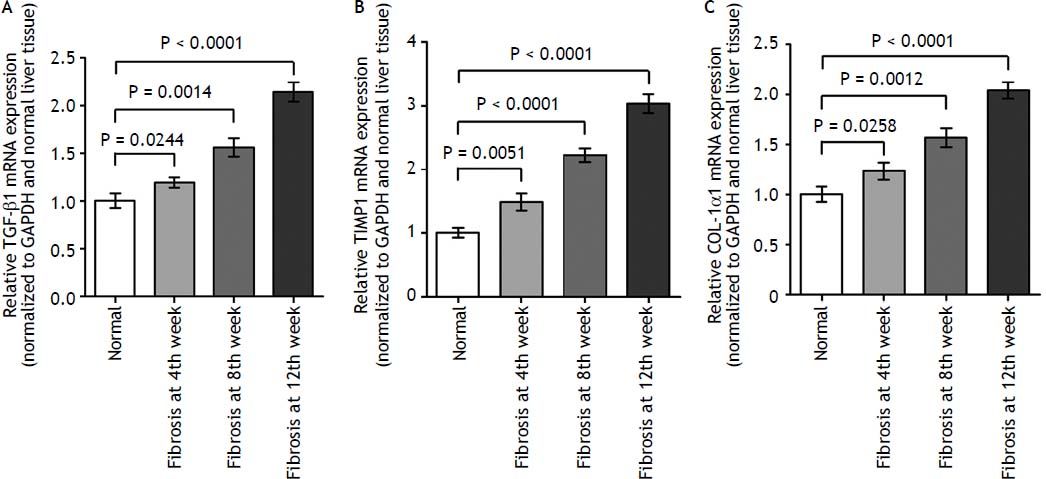
As demonstrated in figure 2, the kinetics change of mRNA expression level of these 3 fibrosis-related genes include TGF-β1, TIMP-1, and COL-1α1 in rat liver tissue was significantly up-regulated in fibrotic liver tissues when compared to the normal liver tissues (p < 0.01).

The expression levels of three fibrosis related genes in fibrotic and normal liver tissues by qRT-PCR. qRT-PCR analysis showed that the kinetics change of expression levels of three fibrosis related genes in fibrotic tissue were higher than that in normal tissue samples. Values are represented as the mean ± SD of 10 animals in each group. **p < 0.01 compared with the normal group.
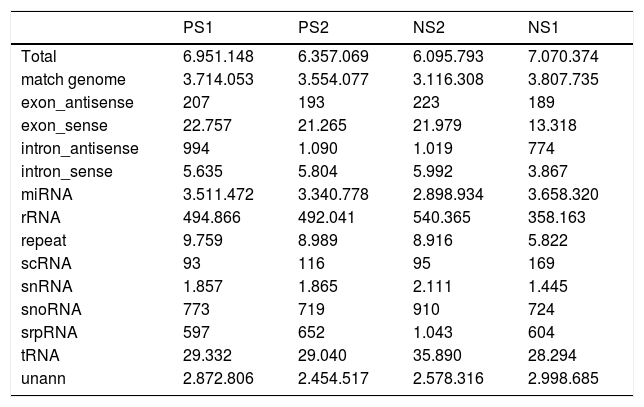
To identify miRNAs that might regulate the progress of PS-induced hepatic fibrosis in rats, deep sequencing technology was employed to sequence four samples, including two fibrosis and two normal liver tissues. After trimming low quality reads, removing adaptors with the aid of a dynamic algorithm and eliminating sequences of length < 18nt, we obtained at least 6.3M and 6.0M clean reads for each fibrosis and normal replicate, respectively. The short oligonucleotide alignment program (SOAP) was used to map all the clean reads to the reference rat genome. When subtracting all reads mapping to annotated miRNAs, rRNA etc., all remaining reads were identified as degraded mRNA by overlap with exons and introns. The results are shown in table 2.
Statistics of reads’ different categories in fibross and normal tissues
| PS1 | PS2 | NS2 | NS1 | |
|---|---|---|---|---|
| Total | 6.951.148 | 6.357.069 | 6.095.793 | 7.070.374 |
| match genome | 3.714.053 | 3.554.077 | 3.116.308 | 3.807.735 |
| exon_antisense | 207 | 193 | 223 | 189 |
| exon_sense | 22.757 | 21.265 | 21.979 | 13.318 |
| intron_antisense | 994 | 1.090 | 1.019 | 774 |
| intron_sense | 5.635 | 5.804 | 5.992 | 3.867 |
| miRNA | 3.511.472 | 3.340.778 | 2.898.934 | 3.658.320 |
| rRNA | 494.866 | 492.041 | 540.365 | 358.163 |
| repeat | 9.759 | 8.989 | 8.916 | 5.822 |
| scRNA | 93 | 116 | 95 | 169 |
| snRNA | 1.857 | 1.865 | 2.111 | 1.445 |
| snoRNA | 773 | 719 | 910 | 724 |
| srpRNA | 597 | 652 | 1.043 | 604 |
| tRNA | 29.332 | 29.040 | 35.890 | 28.294 |
| unann | 2.872.806 | 2.454.517 | 2.578.316 | 2.998.685 |
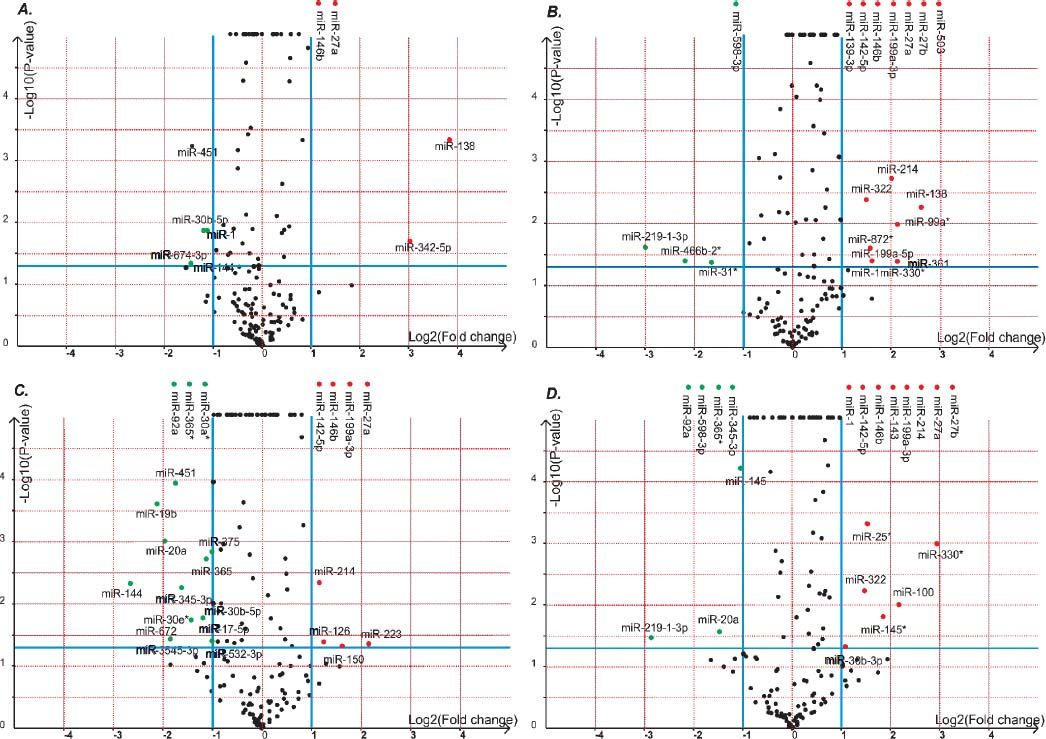
Volcano plot provided comprehensive view about the significance and magnitude of expression alteration of detected miRNAs in each group (Figure 3). In total, 48 miRNAs were identified to be aberrantly expressed at the threshold of |log2Ratio|≥ 1 (P-value ≤ 0.05) in fibrosis tissue compared to normal tissue (Figure 4). Among all these miRNAs, one miRNA (rno-miR-1) showed up-and-down pattern in expression profiles, while the rest of 25 miRNAs were up-regulated and 22 were down-regulatedin at least one group. Interestingly, the consistent expression level of rno-miR-146b and rno-miR-27a suggesting that the role of these common miRNAs may closely related to liver fibrogenesis.

The volcano plot shows the up-regulated and down-regulated miRNAs in four groups. A. NS1 vs. PS1. B. NS1 vs. PS2. C. NS2 vs. PS1. D. NS2 vs. PS2. The horizontal axis represents the fold change between NS and PS. The vertical axis represents the P-value of the t-test for the differences between samples.

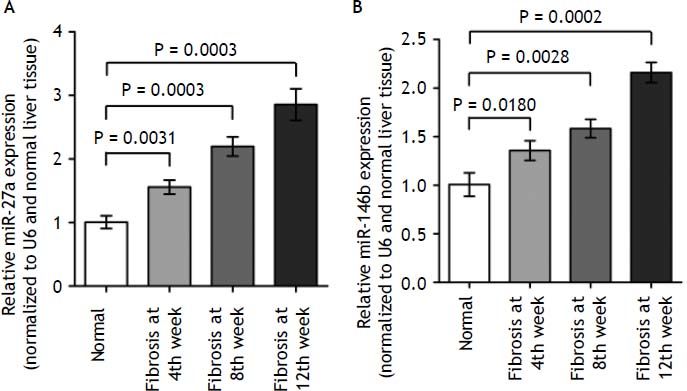
To validate the deep sequencing results, two miRNAs (miR-27a and miR-146b), which steadily expressed across all samples, were selected to be verified by quantitative real-time PCR in all rats. In- deed, we found that miR-27a and miR-146b expression was up-regulated in kinetics change in fibrotic liver tissues when compared to the normal liver tissues (Figure 5, P < 0.01). The qRT-PCR results supported the deep sequencing analysis (Figure 5). Thus, the miRNA expression profiles produced by deep sequencing are reliable.
. Values are represented as the mean ± SD of 10 animals in each group. **p < 0.01 compared with the normal group.")
miR-27a and miR-146b expression in fibrosis and normal liver tissues by qRT-PCR. qRT-PCR analysis showed that the kinetics change of expression levels of miR-27a and miR-146b in the fibrotic and normal tissue samples normalized to U6 and normal liver tissue using the 2ΔΔCT method. miR-27a and miR-146b expression in fibrotic liver tissues when compared to the normal liver tissues (p < 0.01). Values are represented as the mean ± SD of 10 animals in each group. **p < 0.01 compared with the normal group.
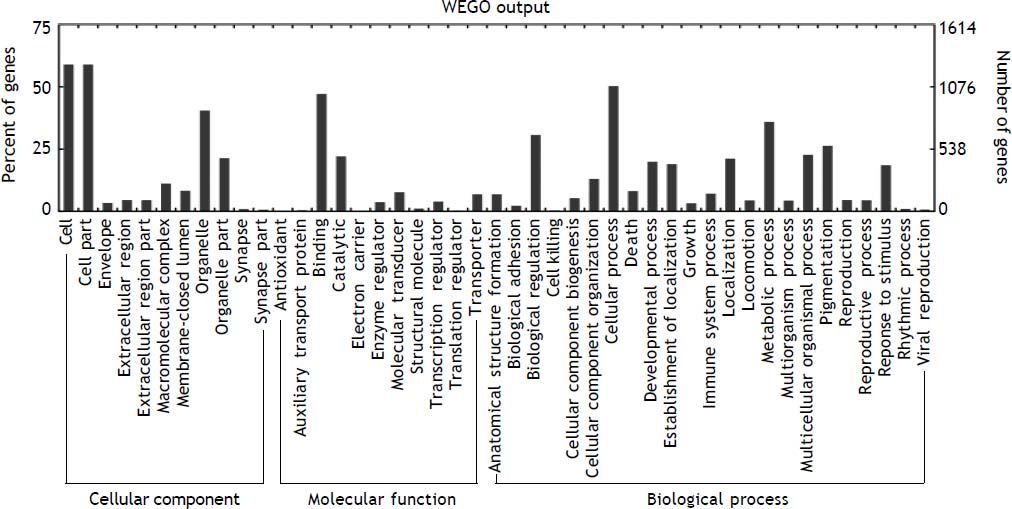
To understand the biological significance of these differentially expressed miRNAs, GO and KEGG pathway analysis was performed to annotate the predicted miRNA target genes into functions and pathways. A total of 2,152 target genes were predicted by 48 deregulated miRNAs, among which 1,378 were assigned at least one GO term (Figure 6). These targets were further classified into functional subcategories. Sequences with GO terms corresponding to the “Cellular Component” group were divided into 11 subcategories, “Molecular Function” into 11 subcategories and “Biological Process” into 23 subcategories. The most abundant GO terms in “Molecular Function” and “Biological Process” were primarily related to “binding” and “cellular process”, making up 47.2 and 50.3% of each subcategory, respectively. The top subcategories found in “Cellular Component” are “cell” and “cell part”, both of which comprised 59.1% of the targets in the subcategory.

Histogram presents of GO terms of target genes. The right y-axis indicates the number of targets in a category. The left y-axis indicates the percentage of a specific category of targets in that main category. All the annotated genes were classified into three major functional categories, namely cellular component, molecular function, and biological process.
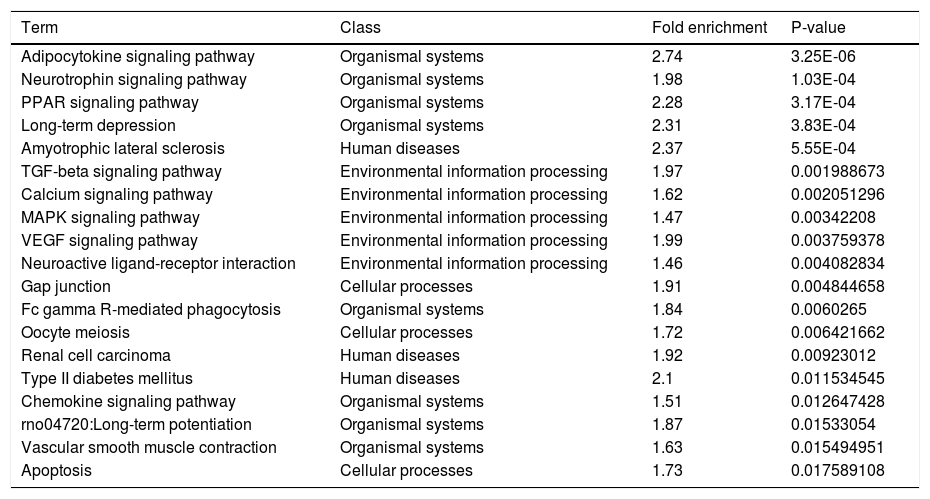
Another functional analysis of KEGG pathway enrichment revealed that 19 pathways were significantly perturbed by a total of 301 annotated target genes (adjusted P-value ≤ 0.05). As listed in table 3, the majority of pathways were tightly associated with organismal systems (Adipocytokine signaling pathway, Neurotrophin signaling pathway, etc.), human diseases (Amyotrophic lateral sclerosis (ALS), etc.) and environmental information processing (TGF-beta signaling pathway, Calcium signaling pathway, etc.). Additionally, we also noticed a significant association in cellular processes, including Gap junction, Oocyte meiosis, Apoptosis etc.
Pathways significantly enriched with target genes predicted by deregulated miRNAs.
| Term | Class | Fold enrichment | P-value |
|---|---|---|---|
| Adipocytokine signaling pathway | Organismal systems | 2.74 | 3.25E-06 |
| Neurotrophin signaling pathway | Organismal systems | 1.98 | 1.03E-04 |
| PPAR signaling pathway | Organismal systems | 2.28 | 3.17E-04 |
| Long-term depression | Organismal systems | 2.31 | 3.83E-04 |
| Amyotrophic lateral sclerosis | Human diseases | 2.37 | 5.55E-04 |
| TGF-beta signaling pathway | Environmental information processing | 1.97 | 0.001988673 |
| Calcium signaling pathway | Environmental information processing | 1.62 | 0.002051296 |
| MAPK signaling pathway | Environmental information processing | 1.47 | 0.00342208 |
| VEGF signaling pathway | Environmental information processing | 1.99 | 0.003759378 |
| Neuroactive ligand-receptor interaction | Environmental information processing | 1.46 | 0.004082834 |
| Gap junction | Cellular processes | 1.91 | 0.004844658 |
| Fc gamma R-mediated phagocytosis | Organismal systems | 1.84 | 0.0060265 |
| Oocyte meiosis | Cellular processes | 1.72 | 0.006421662 |
| Renal cell carcinoma | Human diseases | 1.92 | 0.00923012 |
| Type II diabetes mellitus | Human diseases | 2.1 | 0.011534545 |
| Chemokine signaling pathway | Organismal systems | 1.51 | 0.012647428 |
| rno04720:Long-term potentiation | Organismal systems | 1.87 | 0.01533054 |
| Vascular smooth muscle contraction | Organismal systems | 1.63 | 0.015494951 |
| Apoptosis | Cellular processes | 1.73 | 0.017589108 |
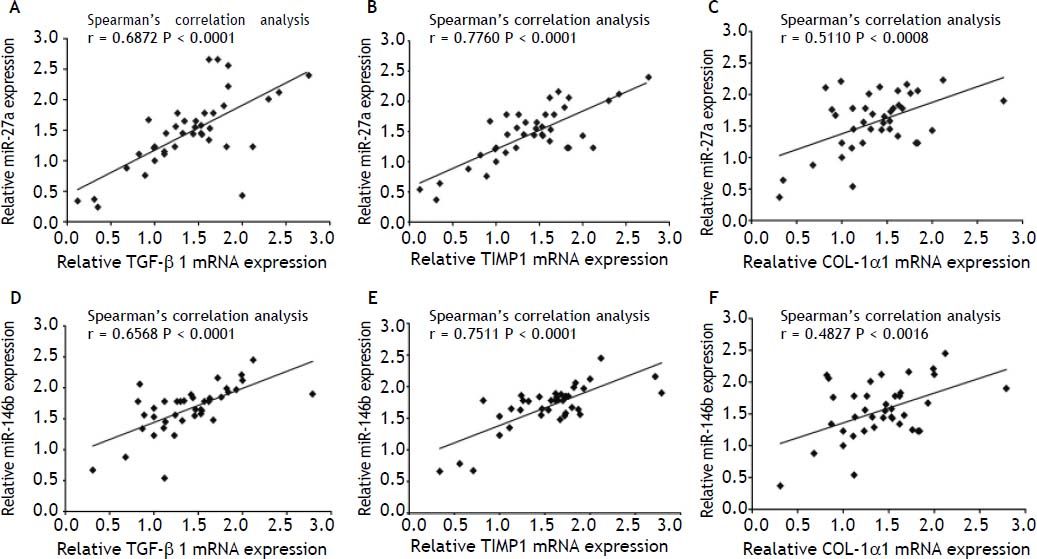
In order to investigate the correlation of these miR-27a and miR-146b with the expression of fibrosis-related genes, we analyzed the relationship between miR-27a, miR-146b and TGF-β1, TIMP1 and COL-1α1 mRNA expression by Pearson’s correlation. Spearman’s correlation analysis showed that the expressions of these 2 miRNAs were strongly and positively correlated with these 3 fibrosis-related genes [r = 0.6872 (miR-27a and TGF-β1), 0.7760 (miR-27a and TIMP-1), 0.5110 (miR-27a and COL-1α1), 0.6568 (miR-146b and TGF-β1), 0.7511 (miR-146b and TIMP-1), 0.4827 (miR-146b p and COL-1α1), P < 0.01] (Figure 7). Thus, our analysis suggested a possible involve- ment of miR-27a and miR-146b in the progression of liver fibrosis.
Discussion
We compared miRNA profiles in PS-induced fibrotic and normal liver by deep sequencing and identified putative targets of differentially expressed miRNAs and enriched pathways with the assistance of bioinformatics. Our comprehensive analysis revealed that the aberrant expression of miRNAs was associated with the progression of liver fibrosis. We identified that 2 highly expressed miRNAs (miR27a and miR-146b) were significantly associated with the progression of liver fibrosis. Coordination of aberrant expression in hepatic miRNAs may contribute to PS-induced hepatic fibrogenesis by regulating several important signaling pathways.
Differentially expressed miRNAs serve as biomarkers of liver fibrotic modelsSeveral miRNA expression profiling by microarrays in the different types of liver fibrosis models have also been carried out.14,15 For example, Li, et al. established a hepatic fibrosis model in rats with dimethylnitrosamine(DMN), array analysis revealed that, as compared with controls, the progression of hepatic fibrosis was associated with up-regulation of 16 miRNAs and down-regulation of 7 miRNAs.14 Roderburg, et al. also found 31 miRNAs were differentially regulated on carbon tetrachloride (CCl4)- induced liver fibrosis, with 10 miRNAs significantly over-expressed in fibrotic livers and 21 miRNAs significantly lower expression when compared with control animals.15 The results in this study reveal that 48 differentially expressed miRNAs in PSinduced liver fibrosis with most of them appear in only a part of the groups.
Among these deregulated miRNAs, it is reported that expression of miR-27b was down regulated in alcoholic mouse fibrosis.34 Recently, dysregulation of several miRNAs such as miR-146b, miR-214, miR-199a-3p, miR-199a-5p, miR-223, miR-342-5p (all up-regulated) as well as miR-92a (down-regulated) was found in DMN-induced liver fibrosis.14 Additionally, miR-199a-3p, miR-199a-5p, miR-223 in CCl4-induced liver fibrosis were also reported to be overexpressed.15 Of note, only three 3 miRNAs (miR199a-3p, miR-199a-5p, miR-223) were commonly up-regulated in the PS-, DMN- and CCl4- induced liver fibrosis models. Intriguingly, the miR-199 families in the human study were positively and significantly correlated to the progressed liver fibrosis,4 suggesting that these miRNAs are involved in different models of liver fibrosis as well as in human livers with advanced liver fibrosis, and might serve as common biomarkers in the diagnosis of hepatic fibrosis.
Regulation of miRNAs miR-27a and miR-146b on rat HSCsEmerging evidence suggests that miRNAs play an essential role in rat HSCs activation.35–37 miR-150 can reduce type I and IV collagen by directly binding to Sp1 and Col4A4 in human LX-2 HSCs.38 Iizuka, et al. found miR-214 was up-regulated in human and mouse livers in fibrosis progression.39 The overexpression of miR-214-5p in LX-2 cells increased the expression of fibrosis-related genes, such as metal-loproteinase MMP-2, MMP-9, α-smooth muscle actin, and TGF-β1.39 In our research, we found miR-27a and miR-146b were up-regulated during hepatic fibrogenesis, and were steadily expressed across all samples. The expression of miR-27a was up-regulated in activated HSCs and influences cell proliferation during HSC activationin the regulation of RXRa expression.40 It has been demonstrated that miR-146b in cardiac fibroblasts played potential role in cardiac fibrosis by targeting MMP-16.41 Another study also demonstrated the overexpression of miR146b in glioblastoma cell inhibited the migration and invasion of malignant gliomas through MMP16 mRNA silencing.42 Our analysis showed that the expressions of miR-146b was strongly and positively correlated with these fibrosis-related genes (TGF-β1,TIMP-1,COL-1a1), and was associated with the progression of hepatic fibrosis progressive,14 suggesting that miR-146b is a potential fibrosis-inducing stimuli in the activated HSC.
MiRNAs appear to be related to liver fibrosis progressIn order to gain further insights into the function of these significantly deregulated miRNAs, GO and KEGG pathway annotation was carried out to annotate the predicted miRNA target genes into functions and pathways. As for our research, target genes were partly enriched in the commonly reportedpathways, such as Gap junction, TGF-beta signaling pathway, apoptosis, etc. Adipocytokine signaling pathway seems to be the most enriched one. Many genes related to HSC activation/liver fibrosis have already been reported, such as gap junctional communication between HSCs is en- hanced after activation by underling complex regulators.43 TGF-beta signaling pathway regulates intercellular communication by affecting the expression level and the phosphorylation state of Cx43 through Snai1 signaling in HSCs.44 Calcium signaling in HSCs was regarded as a crucial molecular basis and took part in hepatic fibrosis.45,46 Previous studies have reported that the mitochondrial pathway of apoptosis plays a significant role in the progression of liver fibrogenesis via HSC activation.35–37 Our data provide evidence that miRNAs may be functional in the hepatic fibrogenesis. However, detailed studies should to be done to uncover the miRNAs function in liver fibrogenesis and its functional mechanism relating to the regulation of liver fibrogenesis.
This is the first study to profile miRNA expression patterns on a genome-wide scale in PS-induced hepatic fibrosis using deep sequencing technology. It should be pointed out that unmeasured differences may exist and may distort the study results. These preliminary data will be confirmed by subsequent studies with larger sample sizes, and more experiments are needed to validate the roles of miRNA and relevant signaling pathways in PS-induced hepatic fibrogenesis. In addition, according to the above mentioned consequence, miR-27a and miR-146b would be potential therapeutic targets during hepatic fibrosis, and this needs more study.
SupportingThe sequences data were submitted to the NCBI Gene Expression Omnibus under accession number GSE54028.
Competing InterestsThe authors declare that they have no competing interests.
AcknowledgmentsWe are greatly indebted to the faculty and staff of the Beijing Genomics Institute and Central South University Xiangya Hospital, whose names were not included in the author list, but who contributed to this program.
Author ContributionsShanfei Ge and Fei Liu carried out the laboratory work and wrote this paper. Xiaowei Wang performed bioinformatics analyses and revised the paper. Xin Yi participated in coordination of the study. Fei Liu collected the samples and assisted with the experiments. Jianping Xie and Fei Liu conceived, designed the study, and revised the paper. All authors have read and approved the final manuscript as submitted, and declare no conflict of interests.



