





Glecaprevir/pibrentasvir is a highly effective and well tolerated treatment for hepatitis C infection. Brazilian patients were not included in the original development studies for glecaprevir/pibrentasvir. This study aimed to assess safety and efficacy of glecaprevir/pibrentasvir in treatment-naïve Brazilian adults without cirrhosis or with compensated cirrhosis.
Patients and methodsEXPEDITION-3 was a Phase 3, open-label, multicenter study in treatment-naïve Brazilian adults with hepatitis C infection genotype 1–6. Patients without cirrhosis (F2 or F3) or with compensated cirrhosis (F4) received 8 or 12 weeks of glecaprevir/pibrentasvir, respectively. The primary efficacy endpoint was the rate of sustained virologic response at post-treatment Week 12. Secondary endpoints were on-treatment virologic failure and relapse rates. Baseline polymorphisms were assessed in NS3 and NS5A. Adverse events and laboratory abnormalities were monitored.
Results100 patients were enrolled, 75 received 8 weeks of treatment and 25 received 12 weeks; all patients completed treatment. Overall sustained virologic response at post-treatment Week 12 rate was high (98.0%; 98/100; 95% confidence interval: 93.0–99.4) and remained high regardless of baseline viral or host factors, including demographics, hepatitis C virus RNA levels, polymorphisms in NS3 and/or NS5A, genotype, and relevant comorbidities. 55% of patients reported ≥1 adverse event, the most common being headache (18.0%). Four patients reported serious adverse events; none were considered drug related or led to study drug discontinuation. No hepatic decompensations were observed.
ConclusionsGlecaprevir/pibrentasvir was effective and well tolerated in treatment-naïve Brazilian patients with hepatitis C infection without cirrhosis and with compensated cirrhosis.
Trial RegistrationClinicalTrials.gov NCT03219216.
In 2015, the World Health Organization (WHO) estimated that 71 million people were living with hepatitis C virus (HCV) [1]. In Brazil, it is estimated that 1.4–1.7 million people are chronically infected with HCV [2]. With such a high prevalence, at the 2017 World Hepatitis Summit in Sao Paulo, the viral hepatitis community requested that viral hepatitis be given the same priority as that given to human immunodeficiency virus, tuberculosis, and malaria, in order to end the epidemic in Brazil [3].
Brazil’s HCV treatment program is growing, with more than 41,000 people having received treatment in 2016, compared with 7500 in 2015 [4]. Furthermore, treatment protocols were also updated in August 2017 to expand treatment eligibility to include patients with F2-stage fibrosis regardless of time of diagnosis, increasing the number of patients eligible for treatment in Brazil to 80,000 [4]. Achieving the WHO’s 2030 elimination targets is considered feasible in Brazil; however, it would require substantial upscaling in prevention activities, screening, and treatment of HCV [4,5]. In particular, increasing the proportion of diagnosed patients and those eligible for treatment is necessary in order to reduce HCV burden in Brazil [6].
Glecaprevir/pibrentasvir, an all oral, pangenotypic, interferon-free, ribavirin-free direct-acting antiviral regimen [7], was granted marketing authorization on the 16th of April 2018 in Brazil [8]. The approved treatment durations for treatment-naïve (TN) patients in Brazil are 8 weeks in those without cirrhosis, and 12 weeks in those with compensated cirrhosis (CC) [9]. Brazilian patients were not included in the original development studies for glecaprevir/pibrentasvir; therefore, this study was conducted to evaluate the efficacy and safety of glecaprevir/pibrentasvir in this specific population. After the initiation of this study, the approved treatment duration was reduced to 8 weeks in all TN patients with CC in the European Union (EU) and United States [7,10]. This change was based on the results of the EXPEDITION-8 study, which was performed to investigate 8 weeks of glecaprevir/pibrentasvir in TN patients with CC [11]. At the time of study design for EXPEDITION-3, the data from EXPEDITION-8 were not yet available.
2Material and methods2.1Study design and patientsEXPEDITION-3 was a Phase 3, open-label, multicenter study in TN Brazilian adults with chronic HCV GT1–6, without cirrhosis or with CC (Fig. 1). A minimum of approximately 35 GT1 and 25 GT3 patients were planned for inclusion across 14 sites (approximately 80 F2–3 patients and approximately 20 F4 patients).
 a minimum of approximately 35 GT1 and 25 GT3 patients and 2) approximately 80 F2–3 and a maximum of approximately 20 F4 patients. G/P, glecaprevir/pibrentasvir; PTW12, post-treatment Week 12; QD, once daily.")
Study schematics.†
†The study enrollment was monitored to meet the following enrollment criteria: 1) a minimum of approximately 35 GT1 and 25 GT3 patients and 2) approximately 80 F2–3 and a maximum of approximately 20 F4 patients. G/P, glecaprevir/pibrentasvir; PTW12, post-treatment Week 12; QD, once daily.
Patients without cirrhosis (F2–F3) were identified by (1) a liver biopsy within 24 months before or during screening that demonstrated the absence of cirrhosis; (2) a FibroScan® (Echosens, Waltham, MA) score of 8.8 to <12.5 kPa within 6 months prior to screening or during screening; or (3) a screening FibroTest (BioPredictive, Paris, France) score of 0.49–0.72 (inclusive). Patients without cirrhosis were enrolled into Arm A and received 8 weeks of treatment. Patients with CC (F4) were identified by (1) a previous histologic diagnosis of cirrhosis on liver biopsy; (2) a FibroScan® score of ≥12.5 kPa within 6 months prior to screening or during screening; or (3) a screening FibroTest score of ≥0.73. Patients with CC were enrolled into Arm B and received 12 weeks of treatment. For enrollment purposes, the result of the liver biopsy superseded those of the FibroScan® and FibroTest, and the results of the FibroScan® superseded that of the FibroTest. Patients with cirrhosis were required to be compensated (Child–Pugh Score of ≤6) at screening. All patients were monitored for 12 weeks after their last dose (post-treatment Week 12 [PTW12]).
2.1.1Key inclusion criteriaEnrolled patients met the following criteria: male or female adults (aged ≥18 years at the time of screening) with chronic HCV GT1–6 infection (mixed and indeterminate GTs were acceptable); positive plasma HCV antibody and HCV RNA viral load ≥1000 IU/mL at screening; documented as without cirrhosis (with a METAVIR equivalent fibrosis stage of F2–F3), or with CC (with a METAVIR equivalent fibrosis stage of F4 and a Child–Pugh score of ≤6). Patients with CC also needed to demonstrate absence of hepatocellular carcinoma (HCC) by a negative ultrasound, computed tomography scan or magnetic resonance imaging within 3 months prior to screening or a negative ultrasound at screening.
2.1.2Key exclusion criteriaPatients were excluded from the study if they met any of the following criteria: current hepatitis B virus (HBV) infection (defined as a positive HBV surface antigen or HBV DNA higher than the lower limit of quantification [LLOQ] in patients with isolated positive anti-HBV core antibody); current or past clinical evidence of Child–Pugh B or C classification (score of >6) or clinical history of liver decompensation, including ascites on physical examination, hepatic encephalopathy, or variceal bleeding; history of solid organ transplantation (unless the transplanted organ had been removed, or was non-functional and the patient was clinically stable off immunosuppressive medication for ≥6 months prior to screening); alanine aminotransferase (ALT) or aspartate aminotransferase (AST) >10 times the upper limit of normal (ULN); total bilirubin >3.0 mg/dL; albumin < lower limit of normal (patients without cirrhosis) or <2.8 mg/dL (patients with CC); platelets <90,000 103/µL (patients without cirrhosis) or <60,000 103/µL (patients with CC); and receipt of any investigational or commercially available anti-HCV agents.
2.1.3EthicsThe study was conducted in accordance with the protocol, International Council for Harmonisation guidelines, applicable regulations and guidelines governing clinical study conduct, and the ethical principles that have their origin in the Declaration of Helsinki. The Independent Ethics Committee/Institutional Review Board reviewed the study prior to initiation and approved the protocol, informed consent and patient information. Informed consent was provided by patients prior to any study-related screening procedures.
2.1.4ProceduresPatients were treated with 300 mg/120 mg glecaprevir/pibrentasvir once daily for 8 weeks in Arm A and for 12 weeks in Arm B. Plasma samples were collected at screening and GTs were assessed using Versant® HCV Genotype Inno LiPA Assay, Version 2.0 or higher (LiPA; Siemens Healthcare Diagnostics, Tarrytown, NY), or by Sanger sequencing of the nonstructural protein (NS) 5B gene region by the central laboratory if the LiPA assay was unable to determine the sample GT. Plasma HCV RNA levels were determined by COBAS® AmpliPrep/COBAS® TaqMan HCV Quantitative Test v2.0 (Roche Molecular Systems, Pleasanton, CA). Study visits took place at screening, Day 1, Week 4, Week 8 (Arm B only), end of treatment (EOT), post-treatment Week 4 (PTW4), and PTW12.
2.2Efficacy endpointsThe primary efficacy endpoint was the percentage of patients who achieved sustained virologic response at PTW12 (SVR12), defined as HCV RNA < LLOQ 12 weeks after the last actual dose of study drug, across all GTs. The secondary efficacy endpoints were the percentage of patients with HCV on-treatment virologic failure (OTVF), defined as (1) a confirmed increase of >1 log10 IU/mL above nadir during treatment or HCV RNA ≥100 IU/mL after HCV RNA < LLOQ during treatment, or (2) HCV RNA ≥ LLOQ at EOT with ≥6 weeks of treatment; and the percentage of patients with HCV virologic relapse, defined as HCV RNA ≥ LLOQ between EOT and 12 weeks after the last dose of glecaprevir/pibrentasvir among patients who completed treatment as planned with HCV RNA < LLOQ at EOT, excluding those who were shown to be reinfected. Completion of treatment for efficacy analyses was defined as any patient with treatment duration ≥52 days and ≥77 days for 8 and 12 weeks, respectively.
2.2.1ResistanceRegions encoding full-length NS3/4A or NS5A were sequenced by next generation sequencing (NGS) from available baseline (BL) samples from all patients. HCV GTs and subtypes were subsequently confirmed via phylogenetic analysis of available NS3/4A and/or NS5A sequences. For resistance analyses, BL polymorphisms (defined as a BL amino acid difference relative to the appropriate subtype-specific reference sequence), and substitutions at the time of virologic failure at amino acid positions of interest for the NS3/4A protease (positions 155, 156, 168) and NS5A inhibitor class (positions 24, 28, 30, 31, 58, 92, 93), relative to subtype-specific reference sequences (detection threshold 15%) were determined by NGS.
2.3Safety assessmentsSafety of glecaprevir/pibrentasvir was assessed by monitoring adverse events (AEs) from Day 1 until 30 days after treatment completion (treatment-emergent period) and laboratory abnormalities during treatment. AEs were coded using the Medical Dictionary for Regulatory Activities (Version 21.1, MedDRA, McLean, VA) and the severity was graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (Version 4.0, Bethesda, MD). AE severity and relationship to glecaprevir/pibrentasvir were determined by the investigator. Clinical laboratory samples were analyzed by a central laboratory. Hepatic laboratory abnormalities were assessed, including ALT values >5 × ULN, total bilirubin >3 × ULN, and post-nadir ALT >3 × ULN with total bilirubin ≤2 × ULN. Safety and laboratory abnormalities were summarized descriptively.
2.4Statistical analysisAnalyses of BL characteristics and efficacy data were performed for the set of all patients in the intention-to-treat (ITT) population, defined as all enrolled patients who received ≥1 dose of the study drug; BL characteristics were also summarized by treatment arm. Sensitivity analyses of the primary efficacy endpoint were performed for the set of all patients in the modified ITT (mITT) population, defined as the ITT population modified to exclude patients who did not achieve SVR12 for reasons other than virologic failure. A backward imputation method was used to impute missing responses for SVR12. For the primary and secondary endpoints, two-sided 95% confidence intervals (CIs) using the Wilson’s score method were provided. Subgroup analyses of the primary endpoint were also performed, with CIs provided for subgroups including ≥10 patients.
Safety analyses were performed for all patients who received ≥1 dose of glecaprevir/pibrentasvir. Treatment-emergent AEs (TEAEs) and laboratory abnormalities were summarized. Statistical Analysis System (SAS Institute, Inc., Cary, NC) Version 9.4 was used for all analyses.
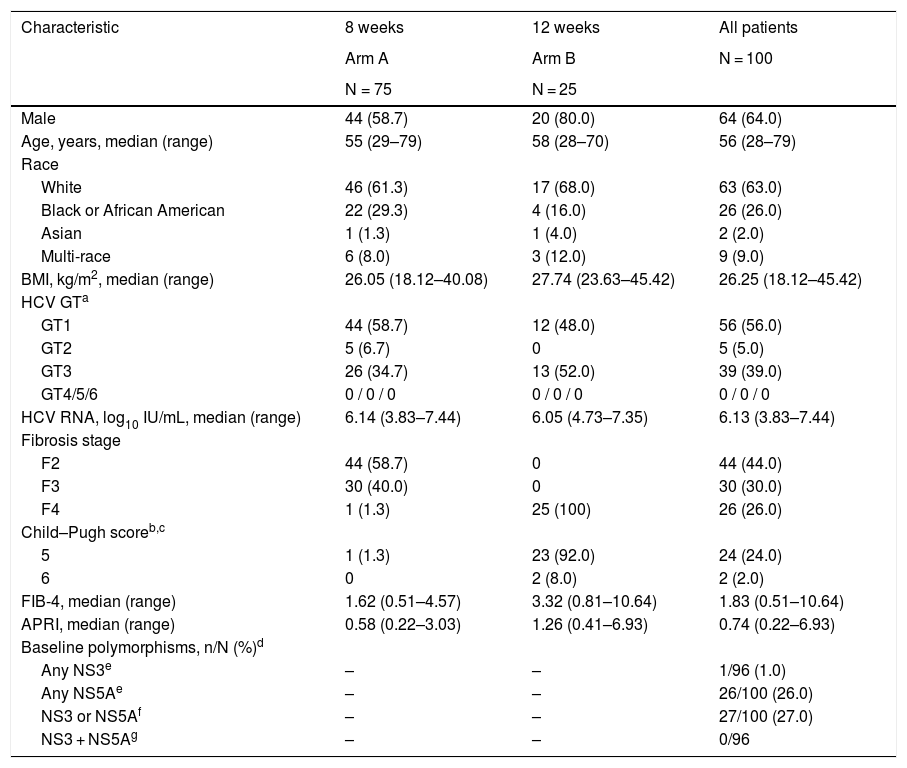
3Results3.1Patient characteristicsA total of 100 patients were enrolled and received ≥1 dose of glecaprevir/pibrentasvir. Seventy-five patients received 8 weeks of treatment (Arm A), and 25 received 12 weeks (Arm B). BL characteristics are summarized in Table 1. All patients completed treatment. All but one patient completed the study; one patient in Arm B was lost to follow-up. Compliance data were available for 94 patients, all of whom were compliant with the treatment.
Baseline demographics and clinical characteristics (ITT population).
| Characteristic | 8 weeks | 12 weeks | All patients |
|---|---|---|---|
| Arm A | Arm B | N = 100 | |
| N = 75 | N = 25 | ||
| Male | 44 (58.7) | 20 (80.0) | 64 (64.0) |
| Age, years, median (range) | 55 (29–79) | 58 (28–70) | 56 (28–79) |
| Race | |||
| White | 46 (61.3) | 17 (68.0) | 63 (63.0) |
| Black or African American | 22 (29.3) | 4 (16.0) | 26 (26.0) |
| Asian | 1 (1.3) | 1 (4.0) | 2 (2.0) |
| Multi-race | 6 (8.0) | 3 (12.0) | 9 (9.0) |
| BMI, kg/m2, median (range) | 26.05 (18.12–40.08) | 27.74 (23.63–45.42) | 26.25 (18.12–45.42) |
| HCV GTa | |||
| GT1 | 44 (58.7) | 12 (48.0) | 56 (56.0) |
| GT2 | 5 (6.7) | 0 | 5 (5.0) |
| GT3 | 26 (34.7) | 13 (52.0) | 39 (39.0) |
| GT4/5/6 | 0 / 0 / 0 | 0 / 0 / 0 | 0 / 0 / 0 |
| HCV RNA, log10 IU/mL, median (range) | 6.14 (3.83–7.44) | 6.05 (4.73–7.35) | 6.13 (3.83–7.44) |
| Fibrosis stage | |||
| F2 | 44 (58.7) | 0 | 44 (44.0) |
| F3 | 30 (40.0) | 0 | 30 (30.0) |
| F4 | 1 (1.3) | 25 (100) | 26 (26.0) |
| Child–Pugh scoreb,c | |||
| 5 | 1 (1.3) | 23 (92.0) | 24 (24.0) |
| 6 | 0 | 2 (8.0) | 2 (2.0) |
| FIB-4, median (range) | 1.62 (0.51–4.57) | 3.32 (0.81–10.64) | 1.83 (0.51–10.64) |
| APRI, median (range) | 0.58 (0.22–3.03) | 1.26 (0.41–6.93) | 0.74 (0.22–6.93) |
| Baseline polymorphisms, n/N (%)d | |||
| Any NS3e | – | – | 1/96 (1.0) |
| Any NS5Ae | – | – | 26/100 (26.0) |
| NS3 or NS5Af | – | – | 27/100 (27.0) |
| NS3 + NS5Ag | – | – | 0/96 |
Data are n (%) unless stated otherwise.
APRI, aspartate aminotransferase to platelet ratio index; BMI, body mass index; FIB-4, fibrosis-4 index for liver fibrosis; GT, genotype; HCV, hepatitis C virus; ITT, intention-to-treat; NS, nonstructural protein; RNA, ribonucleic acid.
Percentages are based on the overall patient population (N) rather than the sub-population of patients with compensated cirrhosis, hence, the sum of percentages across the different categories do not add up to 100%.
Cirrhosis status was based on electronic data capture entries, one patient was enrolled as non-cirrhotic based on a METAVIR score of F2–3, derived from an invalid method for determining cirrhosis. The patient’s valid FibroTest (BioPredictive, Paris, France) result of F4 was used in analysis and a Child–Pugh score was calculated. Therefore, the number of patients reported as cirrhotic does not match the number of patients with a Child–Pugh score result or the number of patients with fibrosis stage F4.
Included are baseline polymorphisms in available samples at amino acid positions 155, 156, and 168 in NS3 and amino acid positions 24, 28, 30, 31, 58, 92, and 93 in NS5A at a detection level of 15%.
“Any” indicates the total number of patients with any polymorphism within the indicated target. Total number of available sequences may vary by target.
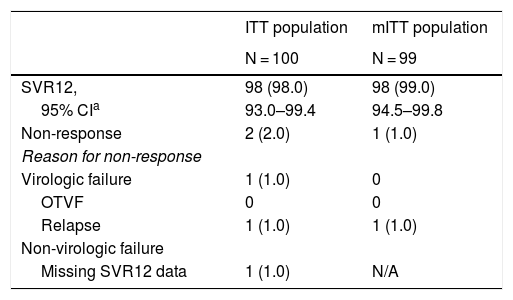
SVR12 was achieved in 98.0% (98/100; 95% CI: 93.0–99.4) of all patients in the ITT population (Table 2). Two patients did not achieve SVR12: one (1.0%; 95% CI: 0.2–5.4) GT3a patient relapsed at PTW4, and one (1.0%) GT3a patient had a non-virologic failure (missing SVR12 data). No patients experienced OTVF (0%; 95% CI: 0.0–3.7); the patient who relapsed was a TN 61-year-old Caucasian female with HCV GT3a infection, a BL fibrosis score of F2, and a BL HCV RNA of 7.44 log10 IU/mL. In the mITT population, the SVR12 rate was 99.0% (98/99; 95% CI: 94.5–99.8). The SVR12 rate was ≥94.9% and ≥97.4% for all GT and fibrosis stage subgroups analyzed in the ITT and mITT populations, respectively (Fig. 2).
SVR12 rates, virologic and non-virologic failure.
| ITT population | mITT population | |
|---|---|---|
| N = 100 | N = 99 | |
| SVR12, | 98 (98.0) | 98 (99.0) |
| 95% CIa | 93.0–99.4 | 94.5–99.8 |
| Non-response | 2 (2.0) | 1 (1.0) |
| Reason for non-response | ||
| Virologic failure | 1 (1.0) | 0 |
| OTVF | 0 | 0 |
| Relapse | 1 (1.0) | 1 (1.0) |
| Non-virologic failure | ||
| Missing SVR12 data | 1 (1.0) | N/A |
Data are n (%) unless stated otherwise.
CI, confidence interval; ITT, intention-to-treat; mITT, modified ITT; N/A, not applicable; OTVF, on-treatment virologic failure; SVR12, sustained virologic response at post-treatment Week 12.
 and the total number of patients in each group. GT, genotype; HCV, hepatitis C virus; ITT, intention-to-treat; mITT, modified ITT; SVR12, sustained virologic response at post-treatment Week 12. a) SVR12 by subgroups ITT. b) SVR12 by subgroups mITT-VF.")
SVR12 rates by subgroups of interest: A. ITT population, B. mITT population.
Numbers represent the number of patients with SVR12 (underlined) and the total number of patients in each group. GT, genotype; HCV, hepatitis C virus; ITT, intention-to-treat; mITT, modified ITT; SVR12, sustained virologic response at post-treatment Week 12. a) SVR12 by subgroups ITT. b) SVR12 by subgroups mITT-VF.
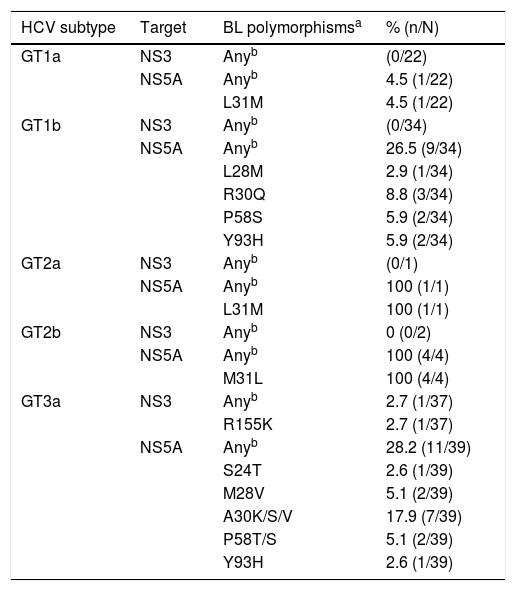
In this study, 56, 5, and 39 patients were infected with GT1, 2, and 3a, respectively. BL polymorphisms in NS3 were rare (1.0%, 1/96), while those in NS5A were detected in 26.0% (26/100) of the patients. BL polymorphisms in NS3 (at positions 155, 156, or 168) were not detected in GT1 and GT2-infected patients and were detected in 2.7% (1/37) of the GT3a-infected patients (Table 3). BL polymorphisms in NS5A (at positions 24, 28, 30, 31, 58, 92, or 93) were detected in 17.9% (10/56), 100% (5/5), and 28.2% (11/39) of the GT1-, GT2-, and GT3-infected patients, respectively (Table 3). Among GT3a-infected patients, three had NS5A-A30K and one had NS5A-Y93H at BL. One GT3a-infected patient experienced virologic failure; at baseline, this patient did not have polymorphisms in NS3 and had A30-K in NS5A. At the time of failure, the patient had treatment-emergent Y93H in NS5A, while NS3 sequence was not available for analysis. BL polymorphisms in NS3 and/or NS5A had no apparent impact on the efficacy of treatment for GT1–3 infected patients.
Baseline polymorphisms in NS3 and/or NS5A.
| HCV subtype | Target | BL polymorphismsa | % (n/N) |
|---|---|---|---|
| GT1a | NS3 | Anyb | (0/22) |
| NS5A | Anyb | 4.5 (1/22) | |
| L31M | 4.5 (1/22) | ||
| GT1b | NS3 | Anyb | (0/34) |
| NS5A | Anyb | 26.5 (9/34) | |
| L28M | 2.9 (1/34) | ||
| R30Q | 8.8 (3/34) | ||
| P58S | 5.9 (2/34) | ||
| Y93H | 5.9 (2/34) | ||
| GT2a | NS3 | Anyb | (0/1) |
| NS5A | Anyb | 100 (1/1) | |
| L31M | 100 (1/1) | ||
| GT2b | NS3 | Anyb | 0 (0/2) |
| NS5A | Anyb | 100 (4/4) | |
| M31L | 100 (4/4) | ||
| GT3a | NS3 | Anyb | 2.7 (1/37) |
| R155K | 2.7 (1/37) | ||
| NS5A | Anyb | 28.2 (11/39) | |
| S24T | 2.6 (1/39) | ||
| M28V | 5.1 (2/39) | ||
| A30K/S/V | 17.9 (7/39) | ||
| P58T/S | 5.1 (2/39) | ||
| Y93H | 2.6 (1/39) |
BL, baseline; GT, genotype; HCV, hepatitis C virus; ITT, intention-to-treat; NS3, nonstructural viral protein 3; NS3, nonstructural viral protein 3; NS5A, nonstructural viral protein 5A.
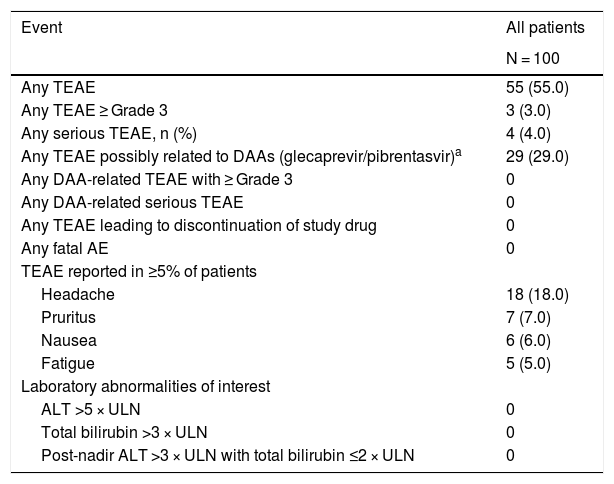
Fifty-five (55.0%) patients experienced one or more TEAEs, most with a maximum severity of Grade 1 (40/55) (Table 4). The most common and only TEAE ≥10% was headache (18.0%). Four patients experienced six serious TEAEs, none of which were considered to be related to glecaprevir/pibrentasvir or led to treatment discontinuation: one patient had a Grade 3 iron deficiency anemia; one patient had a Grade 3 gastric ulcer and a Grade 3 transfusion reaction; one patient had a Grade 4 infectious diarrhea and a Grade 4 acute kidney injury; and one patient had a Grade 1 abdominal pain. No patients experienced a drug-related TEAE of Grade ≥3, discontinued treatment due to an AE, or died during the study. No AEs of special interest (hepatic decompensation/hepatic failure, post-BL events of HCC) were identified. No patients experienced on-treatment hepatic laboratory abnormalities of interest (Table 4).
Summary of adverse events.
| Event | All patients |
|---|---|
| N = 100 | |
| Any TEAE | 55 (55.0) |
| Any TEAE ≥ Grade 3 | 3 (3.0) |
| Any serious TEAE, n (%) | 4 (4.0) |
| Any TEAE possibly related to DAAs (glecaprevir/pibrentasvir)a | 29 (29.0) |
| Any DAA-related TEAE with ≥ Grade 3 | 0 |
| Any DAA-related serious TEAE | 0 |
| Any TEAE leading to discontinuation of study drug | 0 |
| Any fatal AE | 0 |
| TEAE reported in ≥5% of patients | |
| Headache | 18 (18.0) |
| Pruritus | 7 (7.0) |
| Nausea | 6 (6.0) |
| Fatigue | 5 (5.0) |
| Laboratory abnormalities of interest | |
| ALT >5 × ULN | 0 |
| Total bilirubin >3 × ULN | 0 |
| Post-nadir ALT >3 × ULN with total bilirubin ≤2 × ULN | 0 |
Data are n (%).
AE, adverse event; ALT, alanine aminotransferase; DAA, direct-acting antivirals; TEAE, treatment-emergent AE; ULN, upper limit of normal.
In this study of TN Brazilian patients with HCV GT1–6 infection, glecaprevir/pibrentasvir achieved high SVR12 rates of 98.0% in the ITT population and 99.0% in the mITT population. No patients experienced OTVF and one patient relapsed at PTW4. High SVR12 rates were also observed regardless of viral or host factors, including BL polymorphisms in NS3 and/or NS5A, GT, HCV RNA levels and fibrosis stage. Treatment was well tolerated with no new safety signals identified, no serious AEs were considered to be related to glecaprevir/pibrentasvir, and no AEs led to treatment discontinuation. One patient experienced virologic failure, at the time of failure, the patient had treatment-emergent Y93H in NS5A, while NS3 sequence was not available for analysis. Though NS3 sequence at the time of failure was not available for this patient, it should be noted that a pooled resistance analysis of 17 GT3a-infected virologic failures in phase 2 and 3 studies with glecaprevir/pibrentasvir regimen indicated that treatment-emergent substitutions in NS3, specifically Y56H, Q80R, A156G, or Q168(L/R), were detectable in 9 of 17 patients [12].
The high SVR12 rates observed in this Brazilian population are similar to those observed in previous glecaprevir/pibrentasvir clinical studies [13–15]. Available real-world evidence on the use of direct-acting antivirals in Brazil supports their effectiveness in a non-clinical trial setting, and their transferability into real-world populations [16,17]. It should be noted, however, that while this study was conducted using the 12-week regimen for patients with CC, a recent study, EXPEDITION-8, has demonstrated the high efficacy and tolerability of 8 weeks of glecaprevir/pibrentasvir in TN patients with CC for all GTs [11]. These findings have led to the approval of the 8-week regimen for all TN patients in the EU and United States [7,10]. The data from EXPEDITION-8 have also been submitted to the Brazilian Minister of Health (ANVISA) for review. Shorter treatment duration in TN patients has the potential to improve treatment adherence, reduce healthcare-associated costs [11], and may be more convenient for patients [18].
Limitations of the study include the non-comparative, open-label design of the study. No GT5 or GT6 patients were enrolled in the study, reflecting the low prevalence of GT5 and GT6 in Brazil: 0.1% and 0%, respectively [19]. Nonetheless, a similar study in non-Brazilian TN patients (ENDURANCE-5,6) investigated the efficacy and tolerability of 8-week glecaprevir/pibrentasvir in TN patients without cirrhosis and 12 weeks in TN patients with CC and demonstrated 95.7% SVR12 for GT5 (22/23) and 98.4% SVR12 for GT6 (60/61) [15]. Therefore, high SVR12 rates can be expected for GT5 and GT6 patients in the Brazilian population. Furthermore, high SVR12 rates in GT3-infected patients were observed in this study, a generally more challenging GT to treat [20].
5ConclusionIn this study of Brazilian TN patients without cirrhosis or with CC infected with HCV GT1–6, glecaprevir/pibrentasvir demonstrated high efficacy and was well tolerated, with no new safety signals observed. A shorter treatment duration for TN patients with CC (8 weeks), if approved in Brazil, may further support the efforts for HCV treatment in Brazil, ultimately working toward WHO’s HCV elimination target by 2030 [1].AbbreviationsWHO
World Health Organization
HCVhepatitis C virus
TNtreatment-naïve
CCcompensated cirrhosis
EUEuropean Union
GTgenotype
PTW12post-treatment Week 12
RNAribonucleic acid
HCChepatocellular carcinoma
HBVhepatitis B virus
LLOQlower limit of quantification
ALTalanine aminotransferase
ASTaspartate aminotransferase
ULNupper limit of normal
NSnonstructural protein
EOTend of treatment
PTW4post-treatment Week 4
SVR12sustained virologic response at post-treatment Week 12
OTVFon-treatment virologic failure
NGSnext-generation sequencing
BLbaseline
AEadverse event
MedDRAMedical Dictionary for Regulatory Activities
ITTintention-to-treat
mITTmodified ITT
CIconfidence interval
TEAEtreatment-emergent adverse event
Patient consentPrior to any study-related screening procedures being performed on the patient, the informed consent statement was reviewed and signed and dated by the patient, the person who administered the informed consent and any other signatories according to local requirements.
Author contributionsEach author either made substantial contributions to the conception or design of the work, or was involved in the acquisition, analysis, or interpretation of data for the work; and drafted the manuscript or revised it critically for important intellectual content; and provided final approval of the version to be published.
Ethics approvalThe Independent Ethics Committee (IEC)/Institutional Review Board (IRB) reviewed the ethical, scientific, and medical appropriateness of the study before it was conducted. IEC/IRB approval of the protocol, informed consent, and subject information and/or advertising, as relevant, was obtained prior to the authorization of drug shipment to a study site.
Financial supportThe study was designed and conducted by the investigators and the sponsor, AbbVie. The study analysis and financial support for the study were provided by AbbVie. AbbVie participated in the interpretation of data and in the review and approval of the content. All authors had access to all relevant data and participated in writing, review, and approval of the manuscript for publication; the corresponding author had full access to all data and the final responsibility to submit for publication.
Conflict of interestAbbVie sponsored the study (NCT03219216); contributed to its design; and participated in the collection, analysis, and interpretation of the data and in the writing, reviewing, and approval of the publication.
Mario Peribañez-Gonzalez: principle investigator of this study; invited speaker in medical events. Hugo Cheinquer: principle investigator of this study. Lino Rodrigues: employee of AbbVie and may hold stock or share options. Maria Patelli Lima: principle investigator of this study. Mário Reis Álvares-da-Silva: principle investigator of this study; advisory board, research and/or speaker for AbbVie, Bayer, Gilead, Novartis. José Madruga: principle investigator of this study. Edison Roberto Parise: principle investigator of this study. Mário Guimarães Pessoa: principle investigator of this study; scientific advisor, speaker; employee of University São Paulo (state University). Juvencio Furtado: principle investigator of this study. Marcia Villanova: principle investigator of this study. Adalgisa Ferreira: principle investigator of this study. Felipe Mazzoleni: principle investigator of this study. Ecio Nascimento: principle investigator of this study. Giovanni Faria Silva: principle investigator of this study; advisory board, research and/or speaker for AbbVie, Gilead, Bayer, Bristol Myers Squibb. Linda Fredrick: employee of AbbVie and may hold stock or share options. Preethi Krishnan: employee of AbbVie and may hold stock or share options. Margaret Burroughs: employee of AbbVie and may hold stock or share options. Tania Reuter: principle investigator of this study.
AbbVie sponsored the trial (NCT03219216). We thank the patients, trial investigators, coordinators, and trial staff who made this trial possible. Glecaprevir was identified by AbbVie and Enanta. Medical writing support was provided by Annie Massa, MBiolSci, and Jenny Mayes, PhD, of Fishawack Communications Ltd.; funded by AbbVie.



