



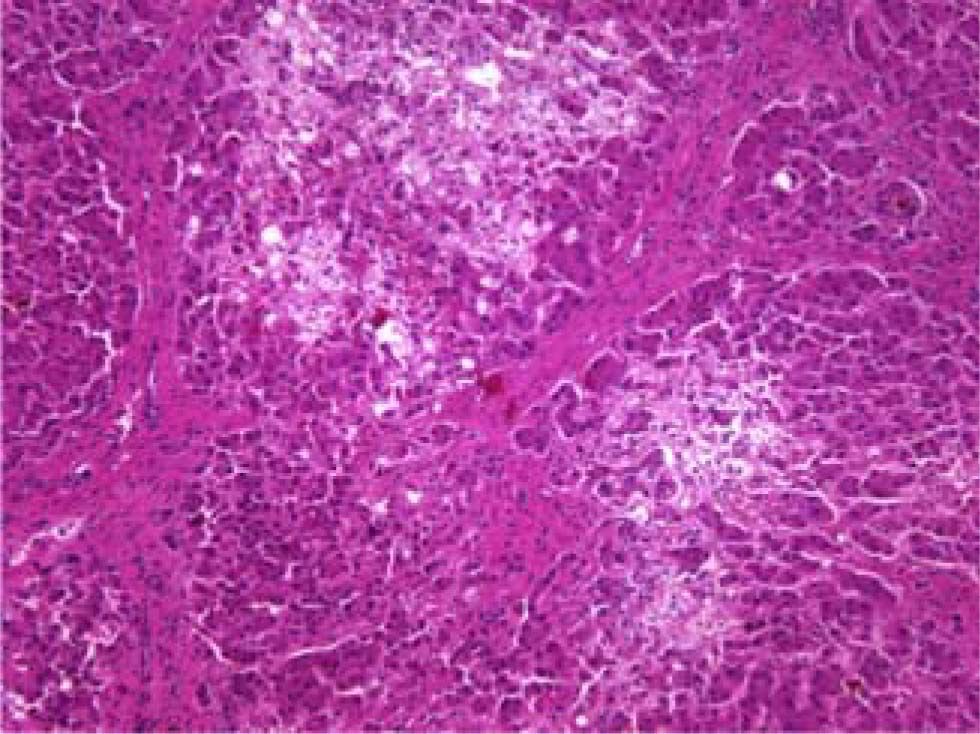
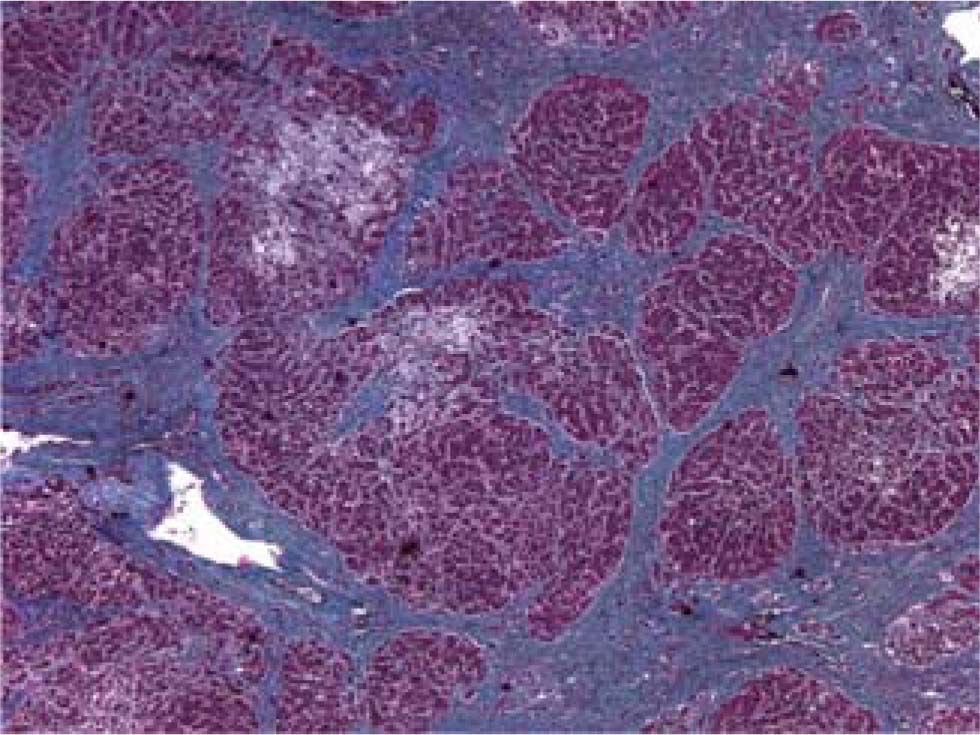
Fibrosing cholestatic hepatitis (FCH) is an aggressive and usually fatal form of viral hepatitis in immunocompromised patients. It is characterized by progressive cholestasis leading to hepatic failure, and a characteristic histopathological features including: periportal fibrosis, ballooning degeneration of hepatocytes, cholestasis, with minimal inflammation. FCH has been reported almost exclusively in heavily immunosuppressed organ transplant recipients or patients with AIDS. This case report describes a previously immunocompetent patient with previously stable chronic hepatitis C who developed fibrosing cholestatic hepatitis after receiving cyclophosphamide and corticosteroids for active glomerulonephritis.
Fibrosing cholestatic hepatitis (FCH) is a rare and severe, progressive form of liver dysfunction that was originally described in patients with recurrent hepatitis B infection after liver transplantation.1 Subsequently, it is reported in patients with other viruses, such as hepatitis C (HCV)2 and in other severely immunosuppressed patients, such as renal and bone marrow allograft recipients and those with the acquired immunodeficiency syndrome (AIDS).2-5 Clinically and biochemically, FCH is characterized by progressive jaundice and liver failure over a period of weeks to months Histologically, it is characterized by periportal and peri-sinusoidal fibrosis, marked ballooning degeneration of hepatocytes, ground glass transformation, prominent cholestasis, neutrophil infiltrates on a background of scant inflammatory infiltration.6,7 Other terms used to describe this entity include «fibrosing cytolytic hepatitis»,8 «steatoviral» and «fibroviral» hepatitis,9 which all draw attention to the hepatocellular degenerative changes, steatosis, and fibrosis that develop despite a paucity of inflammation.9 We report the first case of FCH, outside of transplantation or AIDS, developed in a previously immunocompetent patient with chronic hepatitis C infection that was stable until receiving cyclophosphamide and corticosteroids for treatment of glomerulonephritis.
Case reportClinical historyA 46-year-old Cambodian woman presented with anasarca. She had a six-month history of peripheral edema. Her past medical history was only significant for premature ovarian failure and hypertension. She had no previous surgery or blood transfusion. Furthermore, there was no history of smoking, alcohol, or intravenous drug use. On exam, she had massive ascites and bilateral pitting leg edema. Scalp examination revealed well-circumscribed macules and areas of alopecia. There were also some patches of depigmentation on the scalp, extremities and trunk, and several of these areas were surrounded by inflammatory, scaly, erythematous plaques.
Laboratory investigations revealed a hemoglobin of 65 g/L (115-155), white cell count 6.9 ×109/L (3.8-5.2), platelet count 83 × 109/L (150-400), creatinine 418 umol/ L (40-95), ALT 127 U/L (20-65), AST 10 U/L (10-38), LDH 379 U/L (90-210), albumin 17 g/L (34-50), and INR 1.2. Hepatitis B and HIV screen were negative, but she was found to be hepatitis C positive, genotype 1, with a viral load of 247,000 IU/mL. Serologies for autoimmune disorders, including rheumatoid factor, ANA (anti-nuclear antibodies), dsDNA (double stranded DNA), and anti-ENA (anti-extractable nuclear antigen) were all negative except her complement C3 was mildly decreased at 0.55. Urinalysis revealed hematuria and proteinuria.
Abdominal ultrasound revealed no focal hepatic lesions, biliary duct dilatation or hydronephrosis. A renal biopsy was performed, which revealed active proliferative glomerulonephritis with 20% crescents but no scarring or membranous changes. Immunostaining revealed more IgG than IgM staining. The biopsy was interpreted as typical for lupus nephritis and atypical for hepatitis C associated nephritis. The lupus screen, however, was negative with a negative ANA, DNA, and anti-ENA. Rheumatoid factor and cryoglobulins were also negative. The patient also underwent a transjugular liver core needle biopsy, which showed cirrhosis with a background of chronic hepatitis.
For her acute renal failure, the patient required hemodialysis and was treated with oral cyclophosphamide 150 mg daily and pulse methylprednisolone, followed by high dose of prednisone, 50 mg daily. The cyclophosphamide was discontinued 39 days later because of pancytopenia secondary to bone marrow suppression. However, the patient’s kidney function slowly improved and hemodialysis was discontinued about two and a half month later.
The patient was re-admitted to the hospital four months later with a five-day history of fever and rigors. On exam, she looked unwell. She was febrile at 38°C. Her blood pressure was 165/85, heart rate 90 per minute, respiratory rate 18 per minute, oxygen saturation 92% on 5L of oxygen. Respiratory exam revealed decreased breath sounds bilaterally at the bases with scattered crackles. Abdominal exam showed bulging flanks and shifting dullness, consistent with the presence of ascites. She also had bilateral, pitting edema up to mid-thighs. Laboratory investigations revealed a hemoglobin of 85 g/L (115-155), MCV 85, WBC 6.5 x109/L (3.8-5.2), platelets 16 x 109/L (150-400), sodium 140 mmol/L (135-145), potassium 3.4 mmol/L (3.5-5.0), chloride 107 mmol/L (95-107), bicarbonate 19 mmol/L (22-31), urea 21.9 mmo/L (2.0-8.2), and creatinine 172 umol/L (4095). Her AST was 105 U/L (10-38), ALT 97 U/L (20-65), GGT 400 U/L (10-55), ALP 440 U/L (50-160), total bilirubin 28 umol/L (0-18), LDH 323 U/L (90-210), albumin 13 g/L (34-50), INR 1.56, and PTT 33.8. Septic work-up revealed Klebsiella pneumonia in blood and urine and enterococci bacteremia and appropriate antibiotics were started for her sepsis. Hematology was consulted for anemia and thrombocytopenia. The patient subsequently had bone marrow biopsy, which showed normoblastic erythropoiesis and markedly reduced but morphologically normal megakaryocytes. The patient’s thrombocytopenia was mostly likely multifactorial with a combination of bone marrow suppression related to recent immunosuppressant use as well as sepsis.
Despite intensive treatment, the patient’s clinical condition continued to deteriorate over the next two weeks. Her liver function progressively worsened and she had marked jaundice and ascites. Laboratory investigations revealed persistent thrombocytopenia at 40 × 109/L (150400), Na 139 mmol/L (135-145), K 4.4 mmol/L (3.5-5.0), chloride 109 mmol/L (95-107), bicarbonate 8 mmol/L (22-31), urea 49.6 mmo/L (2.0-8.2), creatinine 171 umol/ L (40-95), GGT 379 U/L (10-55), ALP 1000 U/L (50160), ALT 228 U/L (20-65), AST 254 U/L (10-38), direct bilirubin 583 umol/L (0-18), total bilirubin 710 umol/L (0-18), LDH 456 U/L (90-210), albumin 24 g/L (34-50), INR 1.78, and PTT 27.7.
The patient died shortly thereafter of hepatic failure. At autopsy, the liver exhibited periportal septum formation, cholestasis, fibrosis, limited mixed infiltrate, and hepatocyte ballooning (Figure 1 and Figure 2). The features were consistent with a diagnosis of fibrosing cholestatic hepatitis (FCH).


Fibrosing cholestatic hepatitis has only been described in immunosuppressed patients (patients with HIV, bone marrow or solid organ transplants) with chronic hepatitis B or C. In contrast to the pathogenesis of chronic hepatitis in immunocompetent patients, attributed to cellular immune-mediated hepatocytolysis, FCH has been postulated to result from unimpeded viral replication within hepatocytes, culminating in a direct cytopathic effect, in the setting of immunosuppression.10 Our patient, with chronic hepatitis C, developed rapidly progressive hepatic failure secondary to FCH after immuno-suppressive therapy was initiated.
FCH has a very poor prognosis, with rapid progression to hepatic failure and death occurring within a few months of diagnosis in the majority of patients.1,8 In the setting of FCH that developed after immunosuppression, reduction or withdrawal of immunosuppressive therapy in combination with appropriate anti-viral therapy, may favourably affect the outcome; however, the medical literature with regards to the most optimal therapeutic approach in FCH is still very limited. The postulated direct viral cytopathic effect offers a rationale for the use of antiviral agents to treat this condition, but the role of anti-viral treatment in hepatitis-induced FCH still remains unclear. Tillmann et al. had successfully treated a patient with FCH secondary to HBV with adefovir dipivoxil.11 Similarly, there are a few isolated case reports of FCH with prolonged survival in patients treated with ganciclovir12 and lamivudine.13,14 On the other hand, limited use of interferon in the treatment of HCV-related FCH has not yielded clearly beneficial results.2,15 Because of the rarity of this clinical entity, further studies are still needed to elucidate the role of antiviral therapy in FCH.
The interesting and important aspect of this case lies in the fact that our patient had been immunocompetent with stable chronic hepatitis C before treatment with cyclophosphamide and corticosteroids for glomerulonephritis. Her FCH developed secondary to immunosuppressive therapy with a fatal outcome. Given the rapidly progressive course of her FCH as well as concomitant sepsis and renal failure, it was not possible to commence therapy with peginterferon, however, it is unclear if anti-viral therapy would have altered the dismal outcome given its general lack of success in transplant-associated FCH secondary to HCV. Our experience suggests that FCH can complicate standard immunosuppressive therapy in otherwise immunocom-petent patients with chronic viral hepatitis. Nephrologists and patients with viral hepatitis alike should be aware of the risk of this complication developing in the setting of immunosuppressive therapy with consideration given to possible alteration of the immunosup-pressive protocol
ConclusionFibrosing cholestatic hepatitis is a severe, rapidly progressive form of liver dysfunction seen in patients with hepatitis B or C in the setting of significant immunosuppression. Manifested clinically by jaundice and hepatic failure, FCH is characterized histologically by extensive periportal fibrosis, cholestasis, ballooning degeneration of hepatocytes, and minimal inflammatory cellular infiltrate. FCH reflects a direct hepatocytopathic injury linked to high intrahepatic viral antigen expression. To date, there is no known effective therapeutic intervention that can alter the grave natural history of this condition.



