



Alcoholic liver disease (ALD) is a definition encompassing a spectrum of disorders ranging from simple steatosis to cirrhosis and hepatocellular carcinoma. Excessive alcohol consumption triggers a series of metabolic reactions that affect the liver by inducing lipogenesis, increasing oxidative stress, and causing abnormal inflammatory responses. The metabolic pathways regulating lipids, reactive oxygen species (ROS), and immune system are closely related and in some cases cross-regulate each other. Therefore, it must be taken into account that major genetic and epigenetic abnormalities affecting enzymes involved in one of such pathways can play a pivotal role in ALD pathogenesis. However, recent studies have pointed out how a significant predisposition can also be determined by minor variants, such as relatively common polymorphisms, epigenetic modifications, and microRNA abnormalities. Genetic and epigenetic factors can also affect the progression of liver diseases, promoting fibrogenesis, cirrhosis, and ultimately hepatocellular carcinoma. It is noteworthy that some of these factors, such as some of the cytokines involved in the abnormal inflammatory responses, are shared with non-alcoholic liver disease, while other factors are unique to ALD. The study of the genetic and epigenetic components involved in the liver damages caused by alcohol is crucial to identify individuals with high risk of developing ALD, design personalized protocols for prevention and/or treatment, and select the best molecular targets for new therapies.
Alcohol consumption is responsible for 3.8% of global mortality and 4.6% of disability-adjusted life-years (DA-LYs) lost due to premature death.1 Although the definition of a clear risk threshold for alcohol consumption is still debated, a meta-analysis found increased risks of mortality from liver cirrhosis among men and women drinking 12-24 g of ethanol per day.2 The excessive consumption of alcoholic beverages exposes the liver cells to elevated levels of ethanol, causing a series of damages ultimately leading to a class of clinical conditions identified as alcoholic liver disease (ALD).3
ALD represents a wide spectrum of disorders comprising simple steatosis, alcoholic steatohepatitis (ASH), progressive fibrosis, cirrhosis and the development of hepatocellular carcinoma (HCC).3 The pathogenesis of the liver damage follows a progressive course embracing all these disorders, but exceptions are possible, with patients eventually skipping one of the clinical stages. Up to 90% of heavy drinkers develop steatosis, but only a minority of those with steatosis progress to ASH and 10-20% eventually develop cirrhosis.3,4 There is a dose-response relationship between the volume of alcohol consumed with the risk of ALD, and a population study has proved how subjects who consumed more than 120 g/day had the highest risk of cirrhosis, with a prevalence of 13.5%,5 although it must be considered that the risk of cirrhosis is related to the length of time over which an individual has drunk regularly and not simply to the usual amount consumed.6 However, it should be noted that no study has ever been able to distinguish between the effects of daily consumption from the effects of “binge’” (or compulsive) drinking.
In 2010, the burden of ALD resulted in 493,300 deaths and 14,544,000 DALYs, accounting for 0.9% of all deaths and 0.6% of all DALYs in that year.1,7 These numbers highlight how ALD is the most prevalent cause of advanced liver disease and the leading cause of death among adults with excessive alcohol consumption.1,3 The study of ALD incidence in different populations revealed several biases. In European cohorts, the deaths attributable to alcohol account for 11.0% and 1.8% of men and women, respectively.1,6 The young account for a disproportionate amount of this disease burden, with an alcohol-associated mortality over 10% and 25% of female and male youth, respectively.8 Another important difference was reported by a study on American populations, that showed how individuals of Hispanic origin have a greater risk of developing chronic liver disease than their European and African American counterparts.9
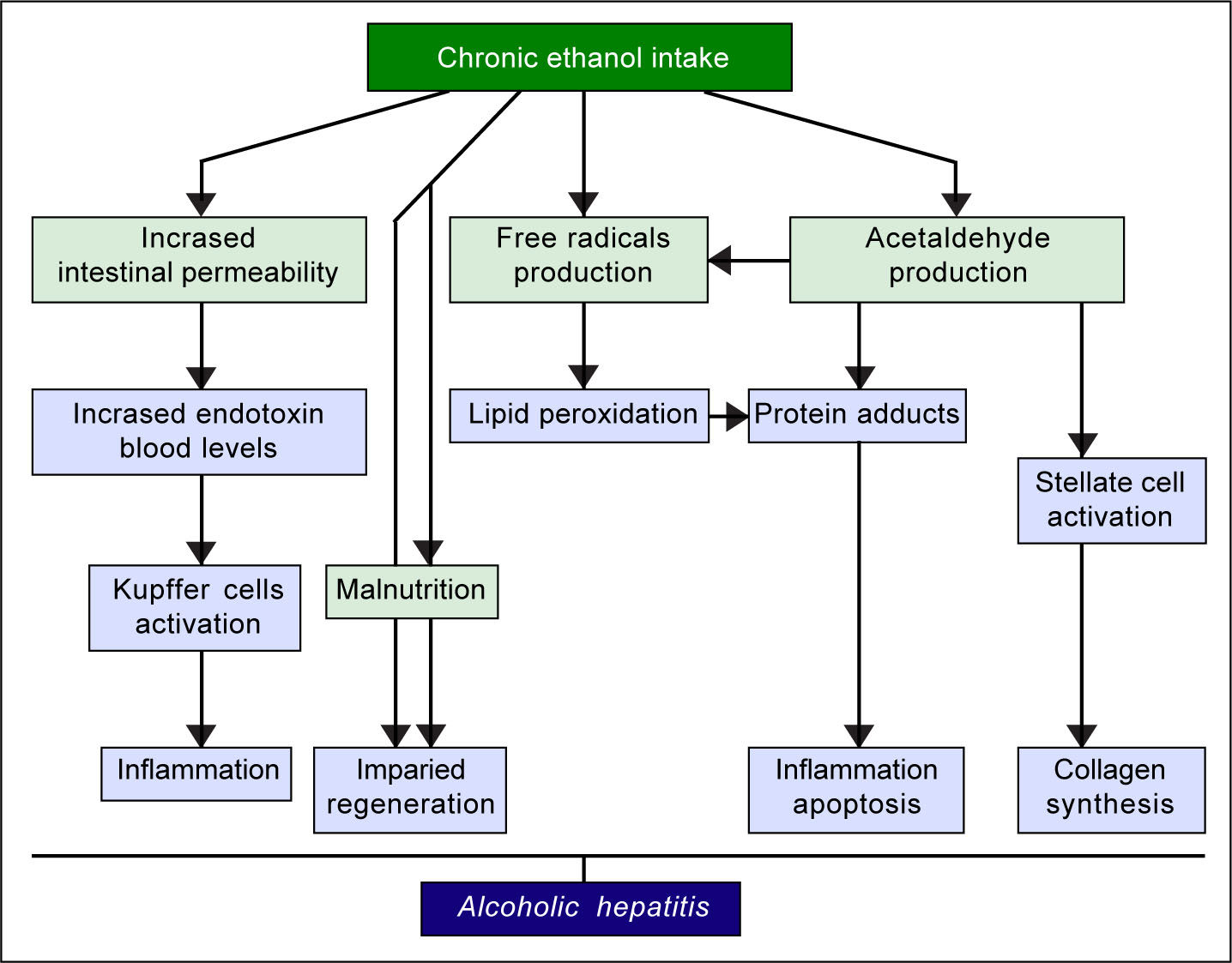
The pathogenic mechanisms underlying ALD involve multiple pathways and seem to converge into determining an inflammatory status in the liver (Figure 1). An increased susceptibility in women as compared to men has been noticed, particularly considering that it requires a lower amount of ethanol in female drinkers to determine liver damage, and suggests that other metabolic and molecular factors, yet to be identified, might play a role in the onset of ALD.10 The information collected by epidemiologic studies indicates how individuals of different ethnicities, exposed to similar environmental contexts, show different ALD incidence, suggesting a potential role for genetic factors. Such role has been confirmed by twin studies, which have revealed a threefold higher disease concordance between monozygotic and dizygotic twins.11,12
Genetic And Epigenetic Mechanisms In Ald
Relevant genetic factors are usually copy number variants (CNVs) or single nucleotide polymorphisms (SNPs), that affect the function of one or more genes in a way that affects the probability of an individual to develop a certain trait, either increasing or decreasing it. Recently, it has been proven how epigenetic factors, can also affect the expression and function of several genes, including several ones involved in liver metabolism and cellular response to alcohol.13 Identification of genetic and epigenetic factors can be important not only for the screening of individuals at risk, but also for the study of the pathogenic mechanisms underlying ALD.
The first evidence supporting the influence of genetic factors on ethanol effects on liver involved the alcohol metabolizing enzymes, alcohol dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH). Sequence polymorphisms in the genes encoding these preoteins have been associated with changes in enzyme kinetics. For example, the SNP rs1229984 in the ADH1B gene is found in 19-91% of East Asians, but in 0-10% of other populations.14 Similarly, the SNP rs671 in ALDH2 is found in 30-50% of East Asians and is basically absent in other populations.15 These variants affect production and removal of the toxic metabolite acetaldehyde, resulting in the development of unpleasant symptoms, such as flushing, nausea, vomiting, tachycardia, hypotension, dyspnea and headache, that ultimately discourage the assumption of alcohol resulting in a protective effect.16 Although to date there is still a lack of large-scale genome-wide association studies (GWAS) for ALD, some other candidate genes have emerged by several studies, such as GABRB1, DRD4, and TH, PECR, ADH1C, LOC100507053, METAP, and PDLIM5.17-20 However, multiple studies suggest that one genetic variant in particular seems to play a pivotal role in the pathogenesis and progression of this spectrum of disorders: the rs738409 SNP in the patatin-like phospholipase domain containing 3 (PNPLA3) gene21-24 (Figure 2). Interestingly, the same variant has also been associated with increased risk of nonalcoholic fatty liver disease and plasma liver enzyme levels by two independent GWAS studies.25,26
. This epigenetic mechanism, based on the activation by phosphorylation of Nrf2, coordinates the cellular response to ROS. When exposed to ethanol, certain microRNAs show a positive correlation in up-regulation of the LPS signal in Kupffer cells, which in turns release the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6. This epigenetic mechanism links alcohol consumption to the inflammatory status in the liver that leads to hepatitis and steatohepatitis.")
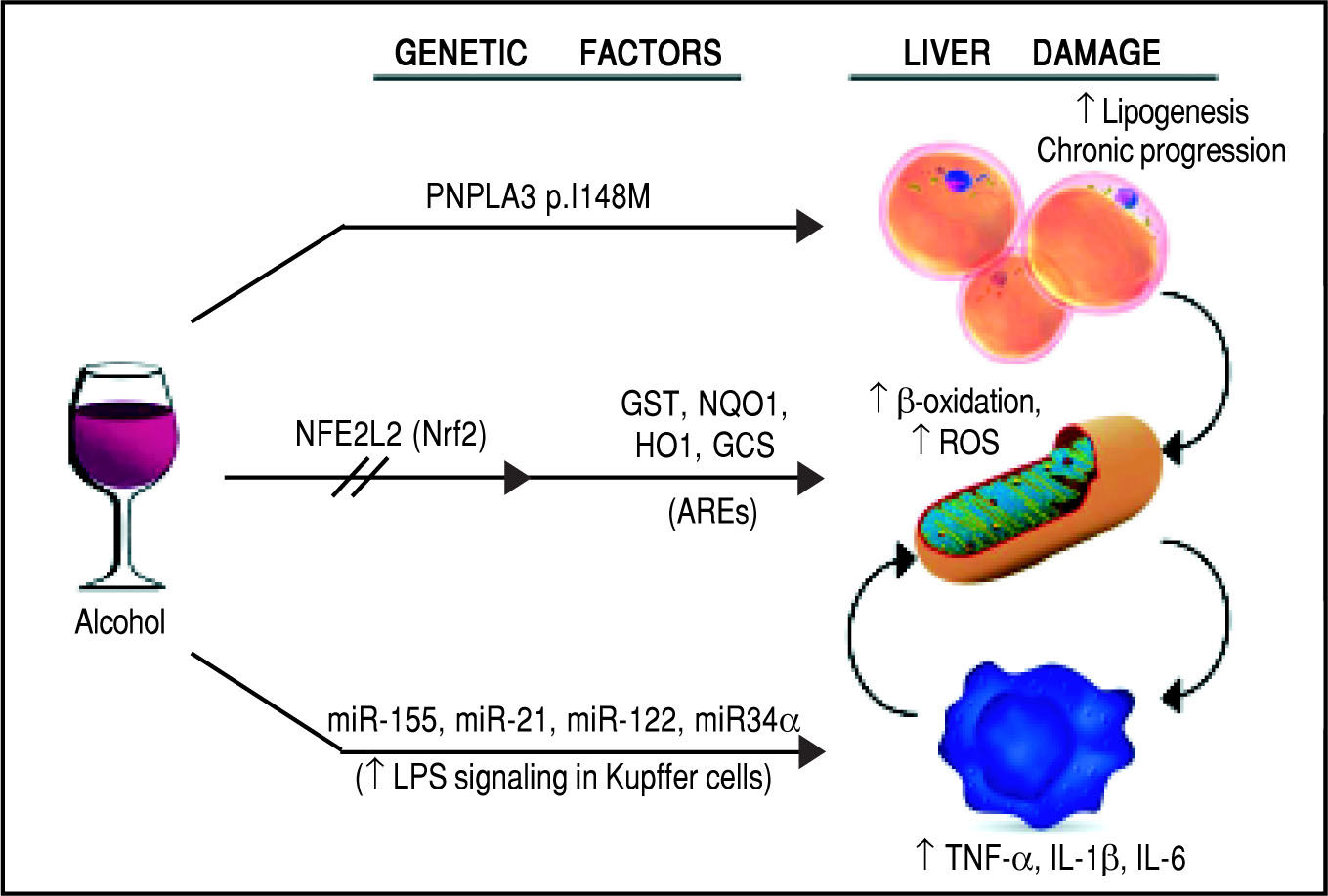
Schematic representation of the genetic and epigenetic mechanisms involved in ALD. The liver damage mediated by ethanol intake, is mediated by three main pathways: lipid metabolism, oxidative stress, and inflammation. The PNPLA3 p.I148M SNP predisposes to increased lipogenesis and facilitates the progression of liver diseases to chronic stages. The NFE2L2 gene, encoding for the Nrf2, is activated by oxidative stress and regulates the activity of several antioxidant response elements (AREs). This epigenetic mechanism, based on the activation by phosphorylation of Nrf2, coordinates the cellular response to ROS. When exposed to ethanol, certain microRNAs show a positive correlation in up-regulation of the LPS signal in Kupffer cells, which in turns release the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6. This epigenetic mechanism links alcohol consumption to the inflammatory status in the liver that leads to hepatitis and steatohepatitis.
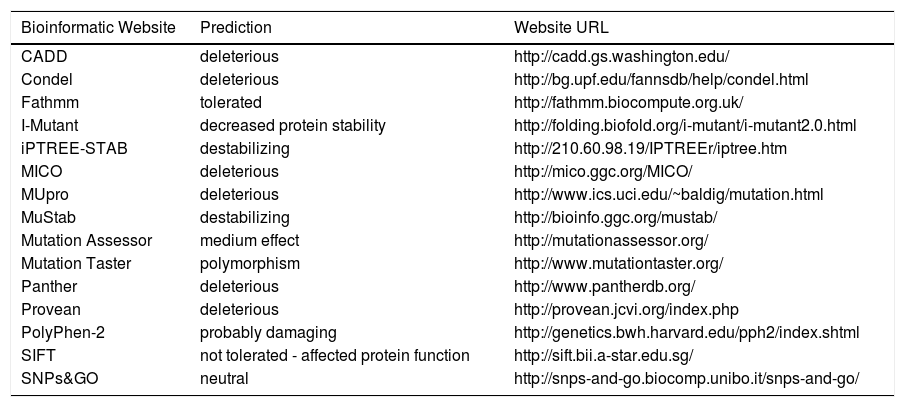
The PNPLA3 gene (OMIM *609567) maps on chromosome 22q13.31, also know as adiponutrin, encodes a 481 amino acid protein with a molecular mass of approximately 53 kDa that in humans is mainly expressed in intracellular membrane fractions in hepatocytes, and is induced in the liver after feeding and during insulin resistance by the master regulator of lipogenesis sterol regulatory element binding protein1c (SREBP1C).27,28 The rs738409 SNP leads to a nucleotide transversion from a cytosine to a guanine at position 444 of the coding region (c.444C > G), in the third of the 9 exons of the gene. This change causes the substitution of the amino acid isoleucine at the residue 148 of the protein with a methionine (p.Ile148 Met or p.I148M). Table 1 lists the predicted effects of the change on the PNPLA3 protein, according to several bioinformatic platforms. Such platforms are designed to create computational models to calculate the impact of an amino acid change, considering parameters like evolutionary conservation of the protein residue and domain, bio-chemical interactions of the original and mutant amino acid, conformational stability of the protein, and frequency of the mutant allele in the general population. Overall, the vast majority of the calls (12 out of 15) indicate a potentially deleterious effect, while only 3 websites consider the p.I148M variant as benign or neutral.
Bioinformatic prediction of the effects of the PNPLA3p.Ile148Met variant.
| Bioinformatic Website | Prediction | Website URL |
|---|---|---|
| CADD | deleterious | http://cadd.gs.washington.edu/ |
| Condel | deleterious | http://bg.upf.edu/fannsdb/help/condel.html |
| Fathmm | tolerated | http://fathmm.biocompute.org.uk/ |
| I-Mutant | decreased protein stability | http://folding.biofold.org/i-mutant/i-mutant2.0.html |
| iPTREE-STAB | destabilizing | http://210.60.98.19/IPTREEr/iptree.htm |
| MICO | deleterious | http://mico.ggc.org/MICO/ |
| MUpro | deleterious | http://www.ics.uci.edu/~baldig/mutation.html |
| MuStab | destabilizing | http://bioinfo.ggc.org/mustab/ |
| Mutation Assessor | medium effect | http://mutationassessor.org/ |
| Mutation Taster | polymorphism | http://www.mutationtaster.org/ |
| Panther | deleterious | http://www.pantherdb.org/ |
| Provean | deleterious | http://provean.jcvi.org/index.php |
| PolyPhen-2 | probably damaging | http://genetics.bwh.harvard.edu/pph2/index.shtml |
| SIFT | not tolerated - affected protein function | http://sift.bii.a-star.edu.sg/ |
| SNPs&GO | neutral | http://snps-and-go.biocomp.unibo.it/snps-and-go/ |
Considering the impact on the protein function, it is relatively surprising how frequent the G allele is in the general population, and even more so in certain ethnicities. According to the 1,000 genomes project (http:// www.internationalgenome.org/home), the G allele has a frequency of 26.2% (1.313 out of 5.008 total alleles tested), with a peak of 71.8% in the Peruvian population (122 out of 170 alleles), and in general a relatively high rate among individuals of Latin American origin (48.4%, 336/694). On the other hand, the subpopulations with the lowest rates of the minor G allele are composed by individuals of African origin (11.8%, 156/1.322 alleles), particularly the Luhya ethnicity (8.6%, 17/198). European individuals also show relatively low rates (22.6%, 227/1.006), with the lowest one detected in the Finnish population (17.2%, 34/198). The allelic frequency is naturally reflected in the genotype distribution: out of 2,508 individuals, 1424 carry the C/C genotype (56.9%), 847 are heterozygous (33.8%), and 233 are homozygous for the G allele (9.3%).
Similar frequencies emerge from other genomic databases, for example the Exome Variant Server (http:// evs.gs.washington.edu/EVS/) reports 2540 G alleles out of 13.006 (19.5%), with a discrepancy between European American (22.1%) and African American individuals (14.5%). The genotype count lists 4,229 individuals with C/C (65%), 2.008 with C/G (30.9%), and 266 with G/G (4.1%). The ExAC Browser Beta database, created by Exome Aggregation Consortium (http://exac.broadinstitute.org/), reports 31.954 G alleles out of 121,386 (26.3%), with peaks of 57.2% in the Latino population (high) and 13.8% in the African one (low).
The fact that the genotype rates overlap the expected frequencies according to the Hardy-Weinberg model (54.5% for C/C, 38.7% for C/G, and 6.9% for G/G, using the 1,000 genome data) suggests that the p.I148M variant does not affect the genetic fitness. In other words, the change does not alter the chances of the carriers to reproduce, and therefore pass the mutant allele to the next generation. This could be explained with the fact that most of the conditions associated with ALD occur after the second-third decade of life and their impact on life quality does not affect the possibility of an individual to procreate.
Wild-type (p.148I) PNPLA3 has lipolytic activity towards triglycerides.27,28 The p.I148M mutation determines a critical aminoacidic substitution next to the catalytic domain, likely reducing the access of substrates and reducing the PNPLA3 enzymatic activity towards glycerolipids, thereby leading to the development of macrovescicular steatosis.27,29 However, other reported a gain of lipogenic function associated with the 148M variant, which would acquire the ability to synthesize phosphatidic acid from lysophosphatidic acid.30,31 In addition, results deriving from murine models gave contradictory results.32-34 Human studies have also suggested a possible direct or indirect influence of PNPLA3 genotype on adipose tissue biology, which however awaits replication.35,36
Other than in ALD, the lipogenic effect of the p.I148M variant appears to play a significant role in predisposing to multiple liver disorders, such as nonalcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH), chronic hepatitis C virus, primary sclerosing cholangitis.37-41 It has also been shown that this substitution can promote disease progression from NAFLD to NASH, fibrosis progression in ALD, and favors hepatic carcinogenesis in several liver diseases.22,42
Oxidative stress and antioxidant response elementsChronic exposure to ethanol increases the production of reactive oxygen species (ROS), lowers cellular levels of antioxidants, and enhances oxidative stress in several tissues, particularly the liver.43 Ethanol is first metabolized in the liver by alcohol dehydrogenase, but when this system reaches saturation, like during chronic alcohol consumption, the cytochrome P450 2E1 (CYP2E1) gets involved, leading to the production of ROS, free radicals, and, most importantly, acetaldehyde, a highly toxic intermediate and carcinogen, which is in turn metabolized by aldehyde dehydrogenase to acetate.44,45 The detoxification process of acetaldehyde is associated to an array of antioxidant mechanisms, regulated by nuclear erythroid 2-related factor 2 (Nrf2), a member of the cap-n-collar basic leucine family of transcription factors.46 In its inactive state, Nrf2 interacts in the cytoplasm with the actin binding protein, Kelch-like ECH-associating protein 1 (Keap1), and is rapidly degraded by the ubiquitin-proteasome pathway. However, upon exposure to oxidative or electrophilic stress, phosphorylation of Nrf2 leads to its dissociation from Keap1, allowing its subsequent translocation into the nucleus.45,47 Once there, Nrf2 binds to antioxidant response elements (AREs), which are particular genomic sequences, that work as targets for molecular messages released by the cells in response to oxidative stress (Figure 2). The Nrf2/AREs functions in partnership with other nuclear proteins, as a strong transcriptional activator of ARE-responsive genes, encoding antioxidant proteins and enzymes such the ones (Table 2): heme oxygenase-1 (HO-1) NAD(P)H; quinone oxidoreductase 1 (NQO1); glutathione-S-transferases (GST); and group C streptococcus (GCS).45
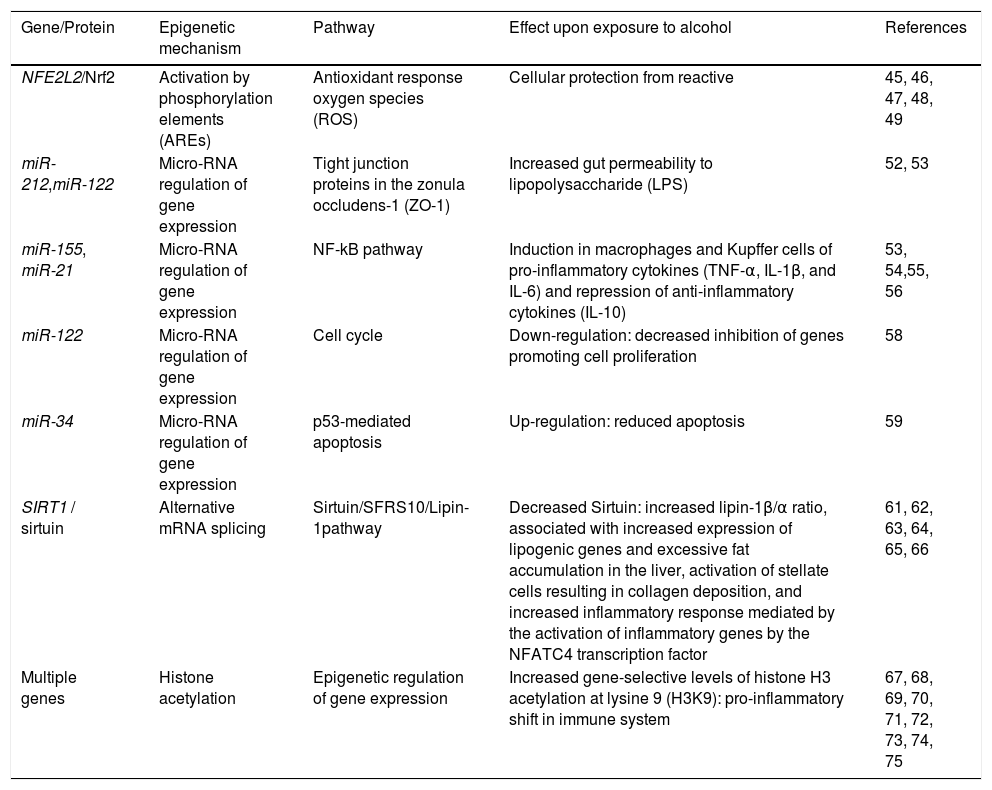
Epigenetic mechanisms involved in ALD.
| Gene/Protein | Epigenetic mechanism | Pathway | Effect upon exposure to alcohol | References |
|---|---|---|---|---|
| NFE2L2/Nrf2 | Activation by phosphorylation elements (AREs) | Antioxidant response oxygen species (ROS) | Cellular protection from reactive | 45, 46, 47, 48, 49 |
| miR-212,miR-122 | Micro-RNA regulation of gene expression | Tight junction proteins in the zonula occludens-1 (ZO-1) | Increased gut permeability to lipopolysaccharide (LPS) | 52, 53 |
| miR-155, miR-21 | Micro-RNA regulation of gene expression | NF-kB pathway | Induction in macrophages and Kupffer cells of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) and repression of anti-inflammatory cytokines (IL-10) | 53, 54,55, 56 |
| miR-122 | Micro-RNA regulation of gene expression | Cell cycle | Down-regulation: decreased inhibition of genes promoting cell proliferation | 58 |
| miR-34 | Micro-RNA regulation of gene expression | p53-mediated apoptosis | Up-regulation: reduced apoptosis | 59 |
| SIRT1 / sirtuin | Alternative mRNA splicing | Sirtuin/SFRS10/Lipin-1pathway | Decreased Sirtuin: increased lipin-1β/α ratio, associated with increased expression of lipogenic genes and excessive fat accumulation in the liver, activation of stellate cells resulting in collagen deposition, and increased inflammatory response mediated by the activation of inflammatory genes by the NFATC4 transcription factor | 61, 62, 63, 64, 65, 66 |
| Multiple genes | Histone acetylation | Epigenetic regulation of gene expression | Increased gene-selective levels of histone H3 acetylation at lysine 9 (H3K9): pro-inflammatory shift in immune system | 67, 68, 69, 70, 71, 72, 73, 74, 75 |
It has also been proven in mouse models that the oxidative stress induced by ethanol via CYP2E1 upregulates Nrf2 activity, which in turn regulates ethanol induction of the cytochrome P450 2A5 (CYP2A5) and protects against alcohol-induced steatosis.48 Chronic ethanol administration in Nrf2-knockout mice significantly increased mortality associated with liver failure.49 The loss of Nrf2 caused a reduced ability to detoxify acetaldehyde, leading to its accumulation, marked steatosis and inflammatory response mediated by Kupffer cells, with consequent depletion of glutathione, one of the most effective antioxidants in the mitochondria. The glutathione deficiency determines an increase in the level of ROS generated by highly energetic processes occurring in the mitochondria, such as the β-oxidation of fatty acids, and ultimately leads to structural and functional changes to mitochondria.49 In order to further prove the key role of the Nrf2/AREs pathway in cellular response to oxidative stress, Wu, et al. demonstrated how activating Nrf2 through Keap1 knockdown and hepatocyte-specific knockout mice blunted the increase in liver levels of serum triglyceride and hepatic-free fatty acid following the exposure to ethanol.50
Inflammation and micro-RNAsAlcohol assumption increases gut permeability and translocation of bacterial products, such as lipopolysaccharide (LPS), also known as endotoxin, into the intestinal and, subsequently, portal circulation.51 Such process involves several genetic and epigenetic effectors: ethanol directly induces an increase in the expression of the miR-212 microRNA, which contributes to the loss of tight junction proteins in the zonula occludens-1 (ZO-1) of the endothelial cells of the intestinal lumen.52 The ZO-1 proteins are also targeted by another microRNA whose expression is influenced by the levels of circulating ethanol, miR-122, and the tight junctions are also weakened by the ethanol metabolite, acetaldehyde53 (Table 2). Once the LPS reaches the liver, it binds to the toll-like receptor 4 (TLR4) on the surface of the Kupffer cells via lipopolysac-charide binding protein (LBP). Such binding activates the Kupffer cells via a signalling cascade that includes CD14, MyD88, MD-2, and finally results in the activation of mi-togen-activated protein kinases (such as ERK1, ERK2, JNK, and p38), NF-KB, and AP-1.54 The binding of LPS to TLR4 induces also an increase in the expression of the microRNAs miR-155 and miR-21, which activate the NF-KB pathway. This cascade effect promotes the production in macrophages, in particular in Kupffer cells, of pro-in-flammatory cytokines, such as tumor necrosis factor (TNF)-oc, interleukin (IL)-1ß, and IL-6, while represses the expression of anti-inflammatory cytokines, such as IL-1053 (Figure 2). It has been proven that microRNAs miR-125b, miR-146a, and miR-155 can regulate the inflammatory response to the TNF-cc induced by LPS in Kupffer cells, and that miR-155 contributes to alcohol-induced activation of TNF-cc in macrophages from patients with ALD.55,56 Abnormal expression levels of several microRNAs have been reported in different liver diseases, generating specific expression profiles for chronic hepatitis, liver fibrosis, and hepatocellular carcinoma.57 If we consider ALD alone, the expression levels of microRNAs, such as miR-155, miR-21, miR-34a, and miR-486, have been reported as increased, other microRNAs, like miR-125b, miR181a, and let-7b, appear to be decreased, while the liver-specific microRNA miR-122 has been reported both as increased and decreased, probably depending on the stage of the disease.53 Two of these microRNAs, miR-122 and miR-34a, produce opposite effects on cell cycle: miR-122 inhibits genes promoting cell proliferation, while miR-34a is a critical regulator of p53-mediated apoptosis.53,58,59 Exposure to ethanol, in both mice and humans, induces up-regulation of miR-34a and down-regulation of miR-122, ultimately promoting the abnormal cell survival and proliferation of hepatocytes observed in ALD53 (Table 2).
One of the target genes of miR-34a is the silent mating type information regulation 2 homolog 1 (SIRT1), encoding the sirtuin protein.60 Sirtuin in involved in a complex pathway regulating β-oxidation of fat acids and lipogenesis, in which the key role is played by lipin-1α. This protein functions both as an Mg2+-dependent phosphatidic acid phosphohydrolase in the triglyceride synthesis pathway and as transcriptional coactivator to promote fat oxidation and suppress de novo lipogenesis.61-63 Lipin-1 is encoded by the LPIN1 gene and has two major isoforms in hepatocytes, depending on an alternative mRNA splicing: lipin-1α, lacking exon 6 and located in the nucleus, and li-pin-1β, containing exon 6, located in the cytoplasm, and associated with increased expression of lipogenic genes and excessive fat accumulation in the liver of animal models.61,64-66 Sirtuin appears to regulate LPIN1 splicing via SFRS10, a member of the SR-like protein family of splicing factors, whose expression is decreased in livers of both mice and humans exposed to high-fat diet.66 Reduced levels of sirtuin have been reported in patients with ALD and the resulting effects have been investigated by Yin, et al. in Sirt1-KO mouse models:61 the decreased expression of SFRS10 causes an increase in the ration between the β and α isoforms of lipin-1, leading to triglyceride accumulation, activation of stellate cells resulting in collagen deposition, and increased inflammatory response mediated by the activation of inflammatory genes by the NFATC4 transcription factor (Table 2). The overall result resembles the histopathological presentation of steatohepatitis in humans, with fatty liver, mild inflammation, and fibrosis. The authors also noted that deletion of Sirt1 exacerbates the effects of chronic-binge ethanol assumption on mouse liver and the oxidative stress in hepatocytes.61
Other than on microRNAs, ethanol has been proven to have several deleterious effects on epigenetic regulation: increased gene-selective levels of histone H3 acetylation at lysine 9 (H3K9) in liver, lungs, spleen, and testes, a phenomenon that seems to be associated with chronic but not acute consumption, increased levels of enzymes mediating histone acetylation, and a generalized increase in DNA methylation.67-73 Once again, these epigenetic-mediated effects of ethanol consumption seem to point primarily to an exacerbation of the inflammatory response, especially considering that a key pro-inflammatory cytokine, such as TNF-α, is silenced by H3K9 methylation and activated by H3K9 acetylation.74 The epigenetic-mediated consequences of chronic alcohol abuse on the immune system affect more than just liver macrophages and Kupffer cells and include effects specific to certain tissues and certain immune cell-types, such as influencing cell recruitment to infected or inflamed tissue, altering cytokine and chemokine production and secretion, skewing differentiation towards a particular cell fate or preventing cell replication, impairing antigen presentation, interfering with phagocytosis and granulopoiesis, or inducing apoptosis.74,75 The ultimate results of ethanol-induced immune dysregulation are increased susceptibility to infection, excessive innate immune response, elevated oxidative stress, and exacerbated or prolonged inflammation due to a skewed macrophage polarization toward the pro-inflammatory M1 phenotype determined by increased production of pro-inflammatory cytokines such as TNF-cc, IL-1ß, IL-6, IFNy, impairment of histone methylation/ acetylation, and promotion of the Th2 lineage speci-fication of the T-helper population of lymphocytes74,75 (Table 2).
In an effort to better characterize the role of innate immune signalling in liver disorders, Petrasek et al. were able to identify differences in ASH versus NASH, as well as pathogenic features shared by both conditions.76 The common traits include a central role played by Kupffer cells, an increase of Gram-negative bacteria in the intestinal lumen, and the beneficial effect of intestinal sterilization on decreasing LPS levels, liver inflammation and fibrosis. In patients with ASH the TLR-4 signalling is mediated via the TRIF/IRF3-dependent and MyD88-inde-pendent pathway, while in NASH cases is mediated by the MyD88-dependent pathway.76 Moreover, while in ASH IL-1ß plays a critical role, the activation of the inflammasome represents an early event and its activation is specific to bone marrow-derived Kupffer cells, in NASH inactivation of IL-1ß protects from steatosis but not from liver damage, the inflammasome is activated later than in ASH and includes hepatocytes in addition to Kupffer cells.76
OverviewThe study of the pathogenic mechanisms in ALD highlights three main pathways in which genetic and/or epigenetic variants can affect an individual's response to ethanol consumption: lipid metabolism, oxidative stress, and immune system. Even if these pathways seem to be quite diverse and distinct for functions, tissues, and substrates, they are connected by an intricate network of tight inter-actions, as proven in several experimental models.54,77 Further studies will be necessary to validate in humans many of the pathogenic mechanisms proved in such experimental models, and it is highly probable that other pathways will be linked to ALD pathogenesis in the future. However, the evidence collected so far is sufficient to justify a prominent role for lipid metabolism, oxidative stress, and immune system in the onset and progression of ALD. Some effects of alcohol consumption are systemic, such as the promotion of the inflammatory response,64 some others are tissue-specific, like the reduced β-oxidation of fatty acids and increased lipogenesis in the liver.21,22,54 Effects occur also in a relatively short time span, like the increased gut permeability to LPS, some others require a multiple-step apparatus and have a later onset, such as the collagen deposition mediated by the activated stellate cells.51 However, it would be a mistake to evaluate each effect individually, even if it affects apparently independent pathways: ethanol operates at the same time on its multiple targets and its effects are often amplified by the cascade effects linking the involved pathways (Figure 2). Both the abnormal lipid metabolism and the pro-inflammatory switch in the immune system increase the production of ROS, which overload mitochondrial activity inducing oxidative stress, leading to tissue damage and ultimately promoting the inflammatory status and the chronic progression to liver disorders from steatosis to steatohepatitis and eventually cirrhosis and fibrosis.22,54,77
Genetic/epigenetic factors can play a role in each of the pathways involved in ALD pathogenesis either by protecting against ethanol effects or exacerbating them. Examples of the first option are the Nrf2/AREs system, regulating the antioxidant response to acetaldehyde, or the SIRT1/SFRS10/Lipin-1 pathway, controlling the expression of lipogenic genes and preventing excessive lipid accumulation in the liver. Constitutive genetic variants can affect proteins involved in these pathways by either decreasing their expression levels and/or disrupting their function. Although there has not been any report yet of patients with ALD carrying loss-of-function mutations in these pathways, it cannot be ruled out the gene dosage effect of multiple variants that are individually considered of unknown significance. Several studies corroborate this hypothesis showing how removing these protective systems in mouse models produce more severe effects after exposure to alcohol.44,49,61 On the other hand, the best documented genetic factor to ALD, the PNPLA3 p.Ile148Met change, represents the typical example of gain-of function variant, in which a carrier does not lack a protective system, but rather possesses a congenital predisposition to amplify the metabolic damage triggered by ethanol.21-24,36 In both cases, in order to better understand the genotype/phenotype correlation in ALD, it is mandatory to improve our knowledge of these pathways and learn how they interact with other cellular functions.
Future PerspectivesIn the era of personalized medicine, the identification of genetic factors predisposing to ALD will have a tremendous impact on the management of this disorder. The most immediate and direct application would be providing biomarkers for the early screening of individuals at risk in the general population to be enrolled in monitoring programs, based on dietary surveillance and/or prophylactic treatment. Genetic screening for the pIle148Met variant in PNPLA3, for example, would be a relative quick and inexpensive service to provide to individuals with positive family history for ALD, and would orient significantly their future clinical management, since this variant can affect not only the onset of the disorder but also its progression.
Another potential application is represented by the identification of potential therapeutic targets. Currently, the treatment of ALD is based on alcohol abstinence, medical treatment (e.g. corticosteroids, prednisolone, pentoxifylline) in the presence of severe liver damage, and liver transplantation in presence of advanced liver disease and/or liver insufficiency.3,78 New approaches have been explored recently and have brought several clinical trials for targeted treatment of alcoholic hepatitis and alcoholic cirrhosis.44 However, a more detailed evaluation of the genetic background will allow the physicians to design the best therapeutic approach for each individual with ALD. For example, targeting the microRNAs involved in the pro-inflammatory switch in macrophages and Kupffer cells will be more efficient and than a steroid-based treatment and will bare less side effects. Moreover, it has to be considered that epigenetic factors can be manipulated by environmental factors, such as diet and medications, and therefore represent an ideal target for genetic therapy.
Including the genetic and epigenetic factors in the design of tailored therapeutic protocols will not only guarantee better results in the treatment of ALD, but will also provide new and more powerful tools for prophylaxis and prevention, resulting in an improvement of both quality and quantity of life for many people.
Abbreviations- •
ADH: alcohol dehydrogenase.
- •
ALD: alcoholic liver disease.
- •
ALDH: acetaldehyde dehydrogenase.
- •
AREs: antioxidant response elements.
- •
ASH: alcoholic steatohepatitis.
- •
CNVs: copy number variants.
- •
CYP2A5: cytochrome P450 2A5.
- •
CYP2E1: cytochrome P450 2E1.
- •
DALYs: disability-adjusted life-years.
- •
GCS: group C streptococcus.
- •
GST: glutathione-S-transferases.
- •
GWAS: genome-wide association studies.
- •
H3K9: histone H3 acetylation at lysine 9.
- •
HCC: epatocellular carcinoma.
- •
IL: interleukin.
- •
Keap1: Kelch-like ECH-associating protein 1.
- •
LBP: lipopolysaccharide binding protein.
- •
LPS: lipopolysaccharide.
- •
NAD(P)H: heme oxygenase-1 (HO-1).
- •
NAFLD: nonalcoholic fatty liver disease.
- •
NASH: nonalcoholic steato-hepatitis.
- •
NQO1: quinone oxidoreductase 1.
- •
Nrf2: nuclear erythroid 2-related factor 2.
- •
PNPLA3: patatin-like phospholipase domain containing 3.
- •
ROS: reactive oxygen species.
- •
SIRT1: silent mating type information regulation 2 homolog 1.
- •
SNPs: single nucleotide polymorphisms.
- •
SREBP1C: sterol regulatory element binding protein-1c.
- •
TLR4: toll-like receptor 4.
- •
TNF-α: tumor necrosis factor-α.
- •
ZO-1: zonula occludens 1.
The authors declare no conflict of interest.
AcknowledgementsThis work is dedicated to the memories of Mr Giuseppe Coloca (1928-2008), and Mrs Carmela Romeo (1929-2010).



