



Background. Hepatotoxicity is a major side effect of treatment with bosentan in patients with pulmonary hypertension (PH). Bosentan is metabolized by the cytochrome CYP2C9 and inhibits the bile salt export pump, which is encoded by ABCB11. This suggests that genetic variants of CYP2C9 and/or ABCB11 may predispose patients to bosentan-induced liver injury.
Material and methods. PH patients with (n = 23) or Without (n = 25) an increase of alanine-aminotransferase (ALT) or aspartate-aminotransferase (AST) during bosentan therapy were included in our analysis. Functionally relevant alleles of CYP2C9 and 16 representative variants of ABCB11 were genotyped. Data were analyzed using logistic regression.
Results. Variants of ABCB11 were not associated with bosentan-induced liver injury. In contrast, variant alleles of CYP2C9 were more common in patients with elevated transaminases (allele frequency 52%) compared to controls (allele frequency 24%, P = 0.04, odds ratio 3.5, 95% confidence interval 1.01-11.8).
Conclusion. Our data indicate hepatotoxicity of bosentan from decreased hepatic metabolism due to common variants of CYP2C9.
Since the advent of drug therapies, the prognosis of pulmonary hypertension (PH) has significantly improved. The specific drug therapies for the treatment of pulmonary arterial hypertension (PAH), one of the five subgroups of PH, target three different signalling cascades - the prostacyclin pathway, the nitric oxide pathway and the endothelin pathway.1,2 Endothelin 1 is the most potent human vasoconstrictor. In addition, it stimulates the proliferation of vascular smooth muscle cells and mediates the development of fibrosis and inflammation.3 Endothelin 1 exerts its effects mainly via the endothelin A and the endothelin B receptors. Bosentan acts as a dual receptor antagonist, which competitively blocks binding of endothelin 1 to both receptors.3
In contrast to other endothelin, bosentan is non-peptidic and contains the structure of a sulphonamide and a bipyrimidine. This compound is orally active with a bioavailability of about 50%. The peak plasma levels are reached within 2-3 h. Bosentan and its metabolites are eliminated predominantly through the liver.
In large clinical trials, bosentan improved exercise capacity, hemodynamics and prognosis in patients with idiopathic PAH (IPAH) and in patients with PAH associated with scleroderma.4,5 In addition, bosentan therapy is effective in various other forms of PAH including Eisenmenger syndrome, portopulmonary hypertension and also in chronic thromboembolic pulmonary hypertension (CTEPH), another subgroup of PH.6–8 However, liver toxicity is a major side effect of bosentan therapy. In a surveillance study, 7.6% of patients treated with bosentan displayed elevated aminotransferase levels and, in almost half of these cases, bosentan therapy had to be permanently withdrawn due to drug induced liver injury.9 Hepatotoxicity caused by bosentan is dose-dependant in higher dosages but also occurs in a subset of patients taking low dosages of bosentan for treatment of PH suggesting an individual genetic predisposition. However, specific genetic risk factors that predispose patients to hepatotoxicity from bosentan therapy are currently unknown.
Following intestinal absorption and hepatic uptake via organic anion transporting polypeptides (OATPs), bosentan is metabolized in hepatocytes by cytochromes CYP2C9 and CYP3A4.10 Subsequently, metabolites are secreted into bile via the organic anion and bilirubin transport protein MRP2, encoded by the ABCC2 gene.11CYP2C9 is polymorphic; the most eminent genetic variants of the gene are the alleles CYP2C9*2, defined by the missense variant c.430C < T (rs1799853) which results in the non-synonymous amino acid substitution p.R144C and CYP2C9*3, defined by the missense variant c.1075A > C (rs1057910) which results in the amino acid substitution p.I359L. Both variants are common in Caucasians with allele frequencies as high as 19%.12 The variant CYP2C9 alleles are associated with lower enzyme activity compared to the wild type allele CYP2C9*1. Homozygotes for the variant alleles are slow metabolizers, whereas heterozygotes are considered intermediate metabolizers for the enzyme’s subtrates, by inference including bosentan.12 In contrast, genetic variation of CYP3A4 in Caucasians is exceedingly rare or was not associated with an effect on drug metabolism in vivo.13
Bosentan and its metabolites inhibit the transport capacity of the hepatocellular bile salt export pump (BSEP), encoded by the ABCB11 gene providing an explanation for cholestatic liver injury from high dosages of bosentan.14 BSEP is a bile salt but not a drug transporting protein. One common variant of the ABCB11 gene (rs228762, p.V444A) leads to lower BSEP expression levels and is associated with drug induced liver injury from various drugs.15 The major transcription factor for ABCB11 is the nuclear bile salt receptor FXR, encoded by the NR1H4 gene.16 A polymorphism of NR1H4 (rs56163822, c.1G/T) was previously confirmed to influence transcription.17 Interestingly, a recent study suggested protection against acetaminophen-induced liver injury from activation of FXR.18
In the current study, we systematically investigated genetic variants of CYP2C9, ABCB11 and NR1H4 as possible genetic risk factors for the development of cholestasis in PH patients during treatment with bosentan.
Material and MethodsPatientsWe included in our study patients with PH and an increase of transaminases (alanine-aminotrans-ferase = ALT and/or aspartate-aminotransferase = AST) following induction of therapy with bosentan. All patients were treated at one of two centres which are spezialized in the treatment of PH (University hospitals Leipzig and Dresden, respectively). Patients with elevated liver function tests during the period from January 2002 to January 2010 who were still alive at the time of analysis were enrolled. The control group was composed of patients without elevation of transaminases during bosentan therapy in the same period.
Twenty-three patients with a confirmed diagnosis of PH of various origins (table) with elevation of aminotransferases (ALT or AST > 1.5 x upper limit of normal) during bosentan therapy and 25 PAH patients without a change of liver enzymes were included (ALT or AST < 1.5 x upper limit of normal). The elevation of aminotransferases of more than 1.5 x upper limit of normal was chosen based on clinical practice in our centers where this elevation routinely resulted in dose modification of the medication. PH was diagnosed according to the published guidelines.2 Other causes of elevation of liver enzymes were excluded: liver ultrasound was performed and viral hepatitis was excluded serologically in all participating subjects. A decompensated right heart failure at the time of measurement of liver enzymes was an exclusion criterion.
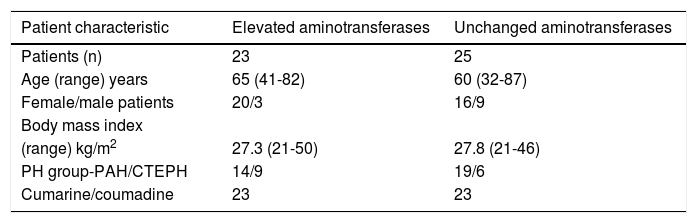
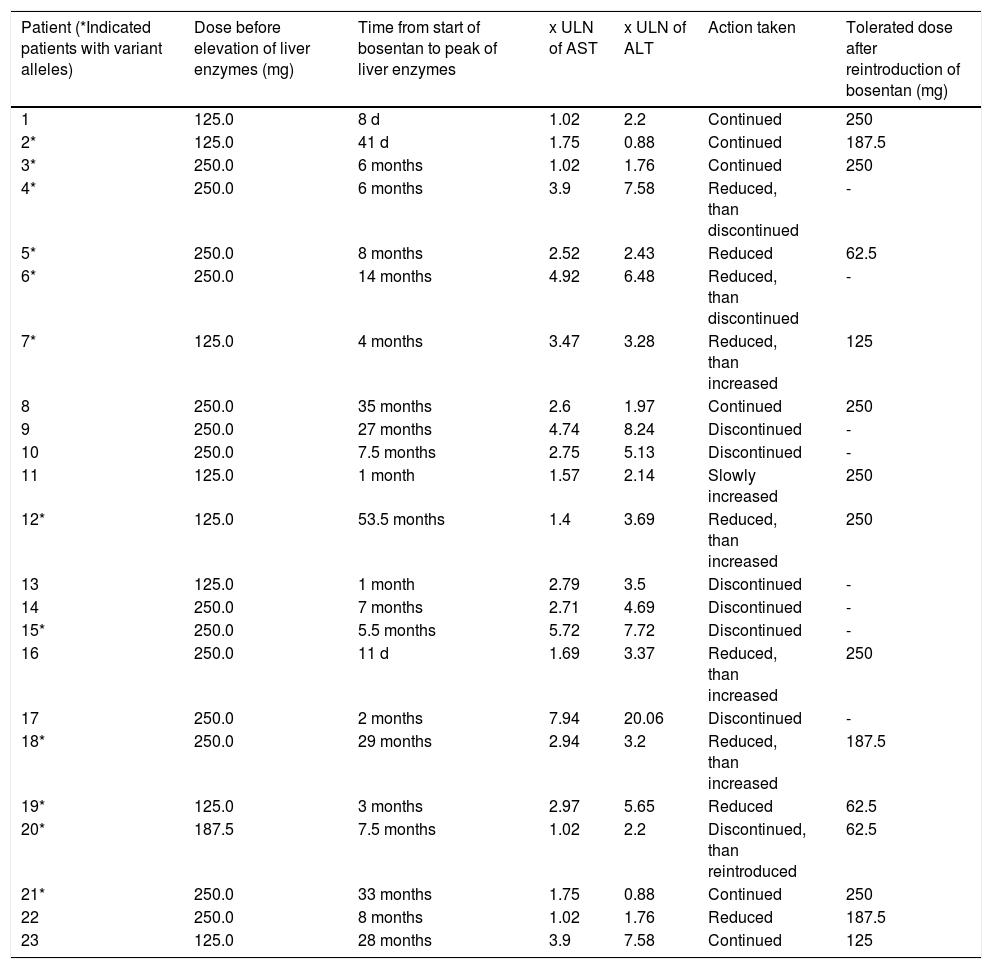
Patient characteristics are summarized in table 1. The sex distribution between the groups of patients with and without elevation of transaminases during therapy with bosentan was slightly skewed. Age, body mass index and distribution to PH subgroups were similar in both groups. All but two patients (both in the control group) took coumarine/couma-dine. Elevation of aminotransferases (ALT or AST > 1.5 x upper limit of normal [ULN]) prior to the treatment with bosentan was associated with right heart failure and occurred only in 2 patients in the control group. Table 2 shows additional information regarding patients with elevated liver enzymes.
Patients characteristics.
| Patient characteristic | Elevated aminotransferases | Unchanged aminotransferases |
|---|---|---|
| Patients (n) | 23 | 25 |
| Age (range) years | 65 (41-82) | 60 (32-87) |
| Female/male patients | 20/3 | 16/9 |
| Body mass index | ||
| (range) kg/m2 | 27.3 (21-50) | 27.8 (21-46) |
| PH group-PAH/CTEPH | 14/9 | 19/6 |
| Cumarine/coumadine | 23 | 23 |
Patients with elevated aminotransferases.
| Patient (*Indicated patients with variant alleles) | Dose before elevation of liver enzymes (mg) | Time from start of bosentan to peak of liver enzymes | x ULN of AST | x ULN of ALT | Action taken | Tolerated dose after reintroduction of bosentan (mg) |
|---|---|---|---|---|---|---|
| 1 | 125.0 | 8 d | 1.02 | 2.2 | Continued | 250 |
| 2* | 125.0 | 41 d | 1.75 | 0.88 | Continued | 187.5 |
| 3* | 250.0 | 6 months | 1.02 | 1.76 | Continued | 250 |
| 4* | 250.0 | 6 months | 3.9 | 7.58 | Reduced, than discontinued | - |
| 5* | 250.0 | 8 months | 2.52 | 2.43 | Reduced | 62.5 |
| 6* | 250.0 | 14 months | 4.92 | 6.48 | Reduced, than discontinued | - |
| 7* | 125.0 | 4 months | 3.47 | 3.28 | Reduced, than increased | 125 |
| 8 | 250.0 | 35 months | 2.6 | 1.97 | Continued | 250 |
| 9 | 250.0 | 27 months | 4.74 | 8.24 | Discontinued | - |
| 10 | 250.0 | 7.5 months | 2.75 | 5.13 | Discontinued | - |
| 11 | 125.0 | 1 month | 1.57 | 2.14 | Slowly increased | 250 |
| 12* | 125.0 | 53.5 months | 1.4 | 3.69 | Reduced, than increased | 250 |
| 13 | 125.0 | 1 month | 2.79 | 3.5 | Discontinued | - |
| 14 | 250.0 | 7 months | 2.71 | 4.69 | Discontinued | - |
| 15* | 250.0 | 5.5 months | 5.72 | 7.72 | Discontinued | - |
| 16 | 250.0 | 11 d | 1.69 | 3.37 | Reduced, than increased | 250 |
| 17 | 250.0 | 2 months | 7.94 | 20.06 | Discontinued | - |
| 18* | 250.0 | 29 months | 2.94 | 3.2 | Reduced, than increased | 187.5 |
| 19* | 125.0 | 3 months | 2.97 | 5.65 | Reduced | 62.5 |
| 20* | 187.5 | 7.5 months | 1.02 | 2.2 | Discontinued, than reintroduced | 62.5 |
| 21* | 250.0 | 33 months | 1.75 | 0.88 | Continued | 250 |
| 22 | 250.0 | 8 months | 1.02 | 1.76 | Reduced | 187.5 |
| 23 | 125.0 | 28 months | 3.9 | 7.58 | Continued | 125 |
The local ethics committee and the responsible authority approved the study protocol. All patients provided written informed consent prior to participation in the study.
GenotypingCommon alleles of CYP2C9 (alleles *2 and *3) were genotyped by melting curve analysis. Variants that cover the common genetic variation of the ABCB11 gene were selected from the HapMap database (www.hapmap.org; selection criteria were r2 > 0.80 and minor allele frequency > 0.05). These tagging single nucleotide polymorphisms (SNPs) as well as the variant rs56163822 of the NR1H4 gene (c.-1G < T) were genotyped in all study participants using the TaqMan SNP Genotyping Assay (Applied Biosystems, Inc., Foster City, CA). The genotyping reaction was amplified on an ABI 2720 Thermal Cycler (Applied Biosystems Inc.; 95 °C for 10 min and 40 cycles with 95 °C for 15 sec and 60 °C for 1 min) and fluorescence was detected on an ABI 7500 RealTime PCR System (Applied Biosystems Inc.).
Statistical analysisAssociations of genetic variants and elevation of transaminases following initiation of bosentan therapy were assessed by binary logistic regression. All analyses were performed in the additive, recessive and dominant models of inheritance. In addition, associations of the ratios of ALT and AST before initiation of bosentan therapy and during treatment and genetic variants were assassed by linary regression analysis. The groups of participants with and without elevation of transaminases differed in the female to male ratio only; in all other parameters they were balanced (Table 1). The differences in sex ratios between groups of patients with elevated transaminases and without an elevation of transaminases during treatment with bosentan were considered a chance finding and, therefore, in the regression analyses no covariates were employed. Correlations between continous variables were assessed by Pearson’s correlation.
P-values < 0.05 were considered to provide nominal evidence for association and are presented without correction for multiple hypothesis testing.
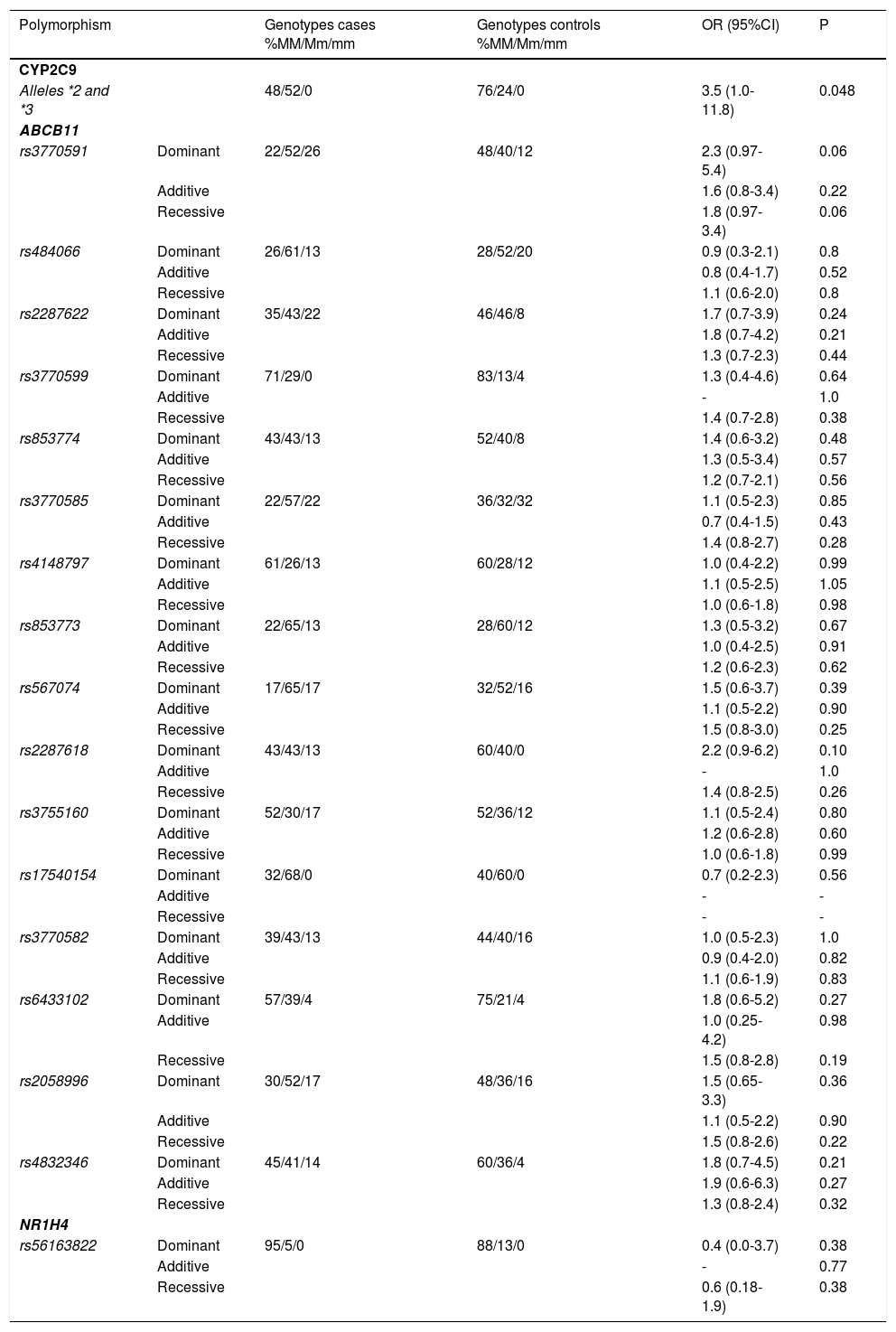
ResultsWe identified 16 SNPs that represented the genetic variation of ABCB11. The analyses of all 16 ABCB11 SNPs in the dominant, additive and recessive models of inheritance did not confirm an association of these variants with the categorical variable “bosentan-induced liver injury” (Table 3). Likewise, variant rs56163822 of the NR1H4 gene was not associated with bosentan-induced liver injury (Table 3). In contrast, alleles CYP2C9*2 and CYP2C9*3 of the CYP2C9 gene were significantly and more than twice as common in PAH patients with an elevation of transaminases (allele frequency 52%) compared to control patients without increased transaminases during bosentan therapy (allele frequency 24%). The odds ratio for experiencing bosentan-induced liver injury was 3.5 for carriers of the CYP2C9*2 and CYP2C9*3 alleles compared with carriers of the wild type allele of the CYP2C9 gene (Table 3).
Results of genotyping.
| Polymorphism | Genotypes cases %MM/Mm/mm | Genotypes controls %MM/Mm/mm | OR (95%CI) | P | |
|---|---|---|---|---|---|
| CYP2C9 | |||||
| Alleles *2 and *3 | 48/52/0 | 76/24/0 | 3.5 (1.0-11.8) | 0.048 | |
| ABCB11 | |||||
| rs3770591 | Dominant | 22/52/26 | 48/40/12 | 2.3 (0.97-5.4) | 0.06 |
| Additive | 1.6 (0.8-3.4) | 0.22 | |||
| Recessive | 1.8 (0.97-3.4) | 0.06 | |||
| rs484066 | Dominant | 26/61/13 | 28/52/20 | 0.9 (0.3-2.1) | 0.8 |
| Additive | 0.8 (0.4-1.7) | 0.52 | |||
| Recessive | 1.1 (0.6-2.0) | 0.8 | |||
| rs2287622 | Dominant | 35/43/22 | 46/46/8 | 1.7 (0.7-3.9) | 0.24 |
| Additive | 1.8 (0.7-4.2) | 0.21 | |||
| Recessive | 1.3 (0.7-2.3) | 0.44 | |||
| rs3770599 | Dominant | 71/29/0 | 83/13/4 | 1.3 (0.4-4.6) | 0.64 |
| Additive | - | 1.0 | |||
| Recessive | 1.4 (0.7-2.8) | 0.38 | |||
| rs853774 | Dominant | 43/43/13 | 52/40/8 | 1.4 (0.6-3.2) | 0.48 |
| Additive | 1.3 (0.5-3.4) | 0.57 | |||
| Recessive | 1.2 (0.7-2.1) | 0.56 | |||
| rs3770585 | Dominant | 22/57/22 | 36/32/32 | 1.1 (0.5-2.3) | 0.85 |
| Additive | 0.7 (0.4-1.5) | 0.43 | |||
| Recessive | 1.4 (0.8-2.7) | 0.28 | |||
| rs4148797 | Dominant | 61/26/13 | 60/28/12 | 1.0 (0.4-2.2) | 0.99 |
| Additive | 1.1 (0.5-2.5) | 1.05 | |||
| Recessive | 1.0 (0.6-1.8) | 0.98 | |||
| rs853773 | Dominant | 22/65/13 | 28/60/12 | 1.3 (0.5-3.2) | 0.67 |
| Additive | 1.0 (0.4-2.5) | 0.91 | |||
| Recessive | 1.2 (0.6-2.3) | 0.62 | |||
| rs567074 | Dominant | 17/65/17 | 32/52/16 | 1.5 (0.6-3.7) | 0.39 |
| Additive | 1.1 (0.5-2.2) | 0.90 | |||
| Recessive | 1.5 (0.8-3.0) | 0.25 | |||
| rs2287618 | Dominant | 43/43/13 | 60/40/0 | 2.2 (0.9-6.2) | 0.10 |
| Additive | - | 1.0 | |||
| Recessive | 1.4 (0.8-2.5) | 0.26 | |||
| rs3755160 | Dominant | 52/30/17 | 52/36/12 | 1.1 (0.5-2.4) | 0.80 |
| Additive | 1.2 (0.6-2.8) | 0.60 | |||
| Recessive | 1.0 (0.6-1.8) | 0.99 | |||
| rs17540154 | Dominant | 32/68/0 | 40/60/0 | 0.7 (0.2-2.3) | 0.56 |
| Additive | - | - | |||
| Recessive | - | - | |||
| rs3770582 | Dominant | 39/43/13 | 44/40/16 | 1.0 (0.5-2.3) | 1.0 |
| Additive | 0.9 (0.4-2.0) | 0.82 | |||
| Recessive | 1.1 (0.6-1.9) | 0.83 | |||
| rs6433102 | Dominant | 57/39/4 | 75/21/4 | 1.8 (0.6-5.2) | 0.27 |
| Additive | 1.0 (0.25-4.2) | 0.98 | |||
| Recessive | 1.5 (0.8-2.8) | 0.19 | |||
| rs2058996 | Dominant | 30/52/17 | 48/36/16 | 1.5 (0.65-3.3) | 0.36 |
| Additive | 1.1 (0.5-2.2) | 0.90 | |||
| Recessive | 1.5 (0.8-2.6) | 0.22 | |||
| rs4832346 | Dominant | 45/41/14 | 60/36/4 | 1.8 (0.7-4.5) | 0.21 |
| Additive | 1.9 (0.6-6.3) | 0.27 | |||
| Recessive | 1.3 (0.8-2.4) | 0.32 | |||
| NR1H4 | |||||
| rs56163822 | Dominant | 95/5/0 | 88/13/0 | 0.4 (0.0-3.7) | 0.38 |
| Additive | - | 0.77 | |||
| Recessive | 0.6 (0.18-1.9) | 0.38 |
In addition, we employed the ratio of AST and ALT before and after initiation of bosentan therapy as a continuous variable of a regression analysis. We identified a high correlation between ratios for AST and ALT before and after initiation of bosentan therapy (Pearson’s correlation coefficient 0.855, p < 0.01). However, these ratios were not affected by genotypes of the CYP2C9 gene (p = 0.41 for AST ratios and p = 0.51 for ALT ratios).
DiscussionThe US Food and Drug Administration suggests considering hepatotoxicity of newly developed drugs if levels of serum aminotransferases are greater than or equal to three times of upper limit.19 However, in a high percentage of patients with lower serum values of aminotransferases, histological assessment of liver biopsies confirmed structural liver damage.20 Therefore, in our study, elevation of liver enzymes was defined as an increase of aminotransferases of more than one and a half times the upper limit of normal, in keeping with common practice in our two centres. However, the majority of cases with elevated liver function tests had an elevation of AST or ALT > 3 x ULN (15/23, 65%).
In this study, alleles CYP2C9*2 and CYP2C9*3 of the CYP2C9 gene were significantly more frequent in patients with elevation of aminotransferases during therapy with bosentan compared with patients without liver toxicity following the initiation of treatment with bosentan. In contrast, genetic variants of the ABCB11 gene, encoding for the bile salt export pump, and NR1H4, encoding its transcription factor FXR were not associated with elevated liver function tests (> 1.5 x ULN). Interestingly, variants of CYP2C9 were not associated with ratios of liver function tests prior to and following initiation of bosentan therapy, suggesting that genetically determined liver toxicity is a categorical and not a continuous trait. Likewise, we also detected no association of variants of ABCB11 and NR1H4 with ratios of liver function tests before and after initiation of therapy. In addition, elevated liver function tests prior to bosentan therapy were not predictive of an increase of liver function tests > 1.5 x ULN after initiation of bosentan therapy (data not shown).
From the results of our systematic genetic analysis, we conclude that patients carrying the variants CYP2C9*2 and CYP2C9*3 of the CYP2C9 gene are exposed to a significantly higher risk of liver toxicity during treatment with bosentan. These variants are associated with a lower activity of the CYP2C9 enzyme,12 resulting in a “bosentan poor metabolizer status.Ú Less efficient metabolization of bosentan, in turn, would result in lower hepatocellular levels of bosentan metabolites such as Ro-48-5033 and higher hepatocellular levels of bosentan itself. Since bosentan is a more potent inhibitor of BSEP compared to its metabolites,14 the “poor metabolizer status” explains, in part, cholestatic liver injury in genetically predisposed individuals. This explanation would provide a compelling link between a genetic association and the physiological mechanism of drug induced liver injury from bosentan. The numbers of male and female patients in the control group reflects the known gender distribution of PAH. Nevertheless, the high incidence of elevation of liver enzymes in female PH patients carrying the genetic variants CYP2C9*2 and CYP2C9*3 remains unclear and was not described previously. Of note, even though carriers of CYP2C9 variants have an odds ratio of 3.5, variation of CYP2C9 does not explain bosentan induced liver injury in all patients. In addition other unknown genetic and non-genetic factors are likely to influence individual risk of liver toxicity. Causality of bosentan treatment and liver injury employing the RUCAM/CIOMS method, the usual way of assessing causality,21 was not formally assessed in the current study. We focussed on genetic aspects of liver injury, but the association of bosentan treatment and hepatotoxicity is generally believed. However, in the current study causality can be assumed but was not proven.
Our results suggest that the examination of genetic variants of CYP2C9 prior to the initiation of bosentan therapy may offer several advantages. In some patients liver toxicity due to bosentan may be preventable. For example homozygous carriers of the CYP2C9 variants (not observed in the current study) may be at risk of severe liver injury. Beyond this potentially important implication, a poor metabolizer of bosentan may need a lower dosage of the drug to avoid liver damage while maintaining treatment efficacy. Since therapy with bosentan is expensive, this may have important economic implications. Moreover, in a large randomized, placebo controlled trial, patients treated with 250 mg bosentan twice daily (twice the standard dose) experienced an additional beneficial effect on exercise capacity.4 Although, this improvement was not statistically significant compared with the standard dose, for some patients increasing the dose of bosentan may be a treatment option. In these rare cases, the determination of the metabolizer status by genotyping CYP2C9 would increase the safety of this potentially harmful therapy. Another option for patients with a “poor bosentan metabolizer status” due to genetic variants CYP2C9*2 and CYP2C9*3 may be the decision to treat with another drug class. This may also increase safety and efficiency of the treatment of PH. Finally, knowledge of the carrier status for CYP2C9 may provide increased safety in the treatment with coumarin anticoagulants, drugs that are also metabolized by CYP2C9 and routinely prescribed in patients with PH.22
The common variant of the ABCB11 gene, rs2287622, and also the variant of the NR1H4 gene, c.-1G < T, are associated with drug induced liver injury caused by various antibiotics, hormonal contraceptives or psychotropics.15 Bosentan and its metabolites inhibit BSEP, resulting in a decreased BSEP-mediated biliary bile salt secretion and putatively in hepatocellular bile salt accumulation.14 Therefore, we investigated these polymorphisms in our cohort as likely determinants of drug toxicity due to bosentan. However, we found no association between polymorphisms of ABCB11 and NR1H4 and liver toxicity in our cohort. Therefore, we conclude that the cholestatic liver injury from bosentan that results in the elevation of liver enzymes is most likely solely caused by the accumulation of bosentan in patients with the “poor metabolizer” status resulting in pronounced inhibition of BSEP, impaired bile salt secretion and decreased bile salt dependant bile flow.
Interestingly, a very recent study from San Francisco with a similar study design found very similar results.23 In this study, the CYP2C9*2 allele was significantly associated with increased transaminases during treatment with bosentan. In contrast, no association was found for CYP2C9*3 and ABCB11 (rs2287622) and drug-induced liver injury during bosentan-treatment. When we designed the study, we decided to combine both the *2 and the *3 allele of CYP2C9 in our analysis since both alleles lead to functionally impaired CYP2C9 enzymes. We re-analyzed our data and confirmed that, in our cohort, it was also the CYP2C9*2 allele that was associated with increased transaminases (p = 0.03), whereas no association was found for the substantially less common *3 allele (p = 0.9). Therefore, the study by Markova confirms our results and indicates that an increased risk of drug toxicity from variant CYP2C9 alleles is not limited to our centres or to European patients.
Our study is limited by the small study population. However, a major advantage of our analysis compared to other studies of drug-induced liver injury is the homogeneous patient cohort and the focus of our study to bosentan only (“one disease, one drug”). In conclusion, our results suggest that the examination of CYP2C9 variants in patients with PH may increase safety and efficiency of drug therapy with bosentan. Thus, the systematic investigation of the genetic background of bosentan metabolism represents an important step in the direction of individualized therapy for PH.
Abbreviations- •
ALT: alanine-aminotransferase.
- •
AST: aspartate-aminotransferase.
- •
BSEP: bile salt export pump.
- •
CTEPH: chronic thromboembolic pulmonary hypertension.
- •
CYP2C9: cytochrome P2C9.
- •
IPAH: idiopathic pulmonary arterial hypertension.
- •
OATP: organic anion transporting polypeptide.
- •
PAH: pulmonary arterial hypertension.
- •
PH: pulmonary hypertension.
- •
SNP: single nucleotide polymorphism.
- •
ULN: upper limit of normal.
Michael Halank and Hans-Jürgen Seyfarth received fees for speaking at conferences and/or consultations from Actelion Pharmaceuticals Deutschland GmbH.



