



This review addresses recent and not so recent works that emphasize on the mechanisms by which liver damage can induce encephalopathy. Hepatic encephalopathy constitutes an intriguing complication in severe liver acute and chronic disease, whose pathophysiology is still not completely understood. In this pathology, alterations in normal brain function are associated with morphological and functional impairments of astrocytes and neurons. A wide spectrum of psychoneurological symptoms has been described and the anatomical substratum is usually associated with brain edema and intracranial hypertension, as well as with changes in the function of brain cells. An increase in blood ammonia, toxic to the brain, depends on the activity of the enzyme glutamine synthetase, the glutamine/glutamate cycle and the brain capacity to eliminate toxic substances. When the concentration of the excitotoxic neurotransmitter glutamate is increased, it acts as a toxic agent, especially when its specific transporters are altered and its uptake is decreased. Glutamine has also been recently considered a toxic substance when its concentration is high, and consequently contributes to brain edema. Finally, the formation of reactive oxygen species, basically produced by mitochondria, influence with their toxic action on membrane lipids, proteins and DNA. In conclusion we suggest that at least these four elements are involved directly in the mechanism of hepatic encephalopathy.
HE: hepatic encephalopathy
CNS: central nervous system
iNOS: inducible nitric oxide synthase
ROS: reactive oxygen species
BBB: blood brain barrier
MSO: methionine-sulfoximine
MPT: mitochondrial permeability transition
GS: glutamine synthetase
OS: oxidative stress
IntroductionHepatic encephalopathy (HE) with subclinical and clinical symptoms constitutes a serious complication in the evolution of acute and chronic hepatic failure. The precise pathophysiological mechanism responsible for HE is not completely understood, and sequential events, cascades and pathways must be clarified before a complete answer is obtained.
Alterations in normal brain function following hepatic injury represent a wide spectrum of psychoneurological symptoms that range from somnolence and excitation to confusion and coma, and are usually caused by brain edema, intracranial hypertension and changes in the function and morphology of brain cells.1
In the early years of the twentieth century, the Russian physiologist Ivan Pavlov described the association of the Eck’s fistula (portacaval anastomosis in dogs) with the presence of ammonia, which shunted into the blood from the intestine,
(Blendis, 2006).1 The pathogenic role of portosystemic shunts and elevated blood ammonia concentrations are part of the hepatic encephalopathy syndrome.
Hepatic encephalopathy, as present in fulminant liver failure, appears in acute form, with rapid evolution to coma, seizures, and a high mortality rate. It usually follows cerebral herniation due to intracranial hypertension and hypoxia. A second form of HE presents a slower onset and milder symptoms and can be reversible. A third form of HE presents a chronic evolution with a prolonged persistence of neuropsychiatry symptoms.2 Another form, a subclinical HE or mild encephalopathy with superficial neuropsychiatric symptoms, which must be recognized to avoid further progression, has also been described.3
Acute and chronic liver failure are associated with many biochemical abnormalities in plasma and brain tissue, which partially explains why the presence of abnormal substances correlate well with the occurrence of memory and behavioral impairments, as observed even with moderate ammonia blood levels in HE.
Among these substances, different concentrations of ammonia, amino acids and neurotransmitters as serotonin, glutamate, glutamine and intermediary metabolites (lactate and pyruvate) can be found.4
In normal subjects, intestinal ammonia, produced from nitrogen products, is taken up by the liver and metabolized to urea. Damaged livers do not accomplish this step adequately, and pathologies as cirrhosis and portal vein thrombosis do not allow splanchnic blood flow to penetrate into the parenchyma; then, portosystemic shunts are created and ammonia and other metabolites are sent to the systemic circulation and finally reach the brain.
In congenital urea cycle disorders, as seen in children with inborn errors of urea cycle enzymes, blood ammonia levels rise soon after birth.
In the hepatic acinus, urea and glutamine metabolism are heterogeneously distributed, being urea synthesis and the enzyme glutaminase periportally localized and glutamine synthetase perivenously localized. Then, the periportal region has a low affinity but a high capacity for ammonia detoxification. Liver glutamine synthetase acts as a scavenger agent for ammonia, and when liver insuffiency is present, a diminished scavenger capacity takes place.
Brain tissue does not have an effective urea cycle, and then the glutamine synthetase from astrocytes is in charge of ammonia and other toxic substances detoxification, as well.
The key role of ammoniaSome years ago, we observed that not always blood ammonia levels correlate with the degree of psychoneurological symptoms in liver cirrhosis. As a result of these observations we decided to use the brain isoenzyme of creatinine kinase as an approximate marker of brain impairment.5 Our results indicated a good relationship between liver failure and the blood concentration of this isoenzyme.
Abou-Assi et al.2 reported that, in some cases, there is a lack of correlation between blood ammonia levels and stages of HE, presuming that there are other factors that participate in HE pathophysiology.
Forty per cent of ammonia is generated by the intestine from nitrogenate substances, caused by the action of bacterial urease and amino acid oxidases, and the other 60% is derived from the metabolism of glutamine and the transamination of other amino acids.
Normally, the efficiency by which liver removes intestinal ammonia maintains it at a low blood concentration. Besides, ammonia is generated as a metabolite product in other cells, especially in muscles. It must be recalled that muscles may remove ammonia ions from blood, changing its plasma concentration. The reactions that are catalyzed by the enzyme glutamate dehydrogenase, glutaminase and AMP-deaminase generate important amounts of ammonia.6
Ammonia either enters the brain by diffusion from the blood or cerebrospinal fluid or is formed in situ from the metabolism of endogenous nitrogen-containing substances. Its excess is toxic to the central nervous system (CNS). As the brain does not produce urea from ammonia, its removal relies almost exclusively on glutamine synthetase, basically localized in astrocytes.7
It has been demonstrated in several studies that ammonia ions participate actively in neuron-astrocyte metabolic interaction, specifically in the functioning of the glutamate/glutamine cycle.6
The morphopathological findings in HE and other hyperammonemic conditions are localized in astrocytes rather than in neurons. The Alzheimer type II astrocyte change is the distinctive morphological alteration in brain of cirrhotic astrocytes, hyperammonemic congenical pathologies and urea cycle disorders.8
The experimental findings by Cummins et al. are, to a certain extent, similar to our results obtained with our model of encephalopathy and prehepatic portal hypertension, with low ammonia concentration and brain damage.9
Ammonia toxic effects act inhibiting the tricarboxylic acid cycle enzymes, •-ketoglutarate dehydrogenase (in vitro)10 and stimulate phospho-fructokinase and other glycolytic enzymes in brain tissue.
Ammonia inhibits excitatory post-synaptic potentials, thereby, producing a general depression of the CNS function; and high levels produce brain energy failure due to its inhibitory action on key tricarboxylic acid cycle enzymes.
The metabolism of ammonia to glutamine appears to be necessary, and is followed by an osmotic disturbance in the brain, mitochondrial dysfunction with oxidative stress, and alterations in brain glucose metabolism. Cerebral blood flow is also altered and strongly influences the development of brain edema and intracranial hypertension. Added factors, such as systemic inflammation, alterations in the brain extracellular concentration of amino acids and neurotransmitters, may contribute to the cerebral alterations in acute liver failure.11
Elevated brain ammonia level is an important etiologic factor in astrocyte swelling and brain edema, the main neuropathological findings in the acute form of hepatic encephalopathy. One factor known to be activated by oxidative/nitrosative stress and mitogen-activated protein kinases (both considered important elements in astrocyte swelling) is nuclear factor kappaB a transcription factor that activates the inducible nitric oxide synthase (iNOS) and other genes. Nitric oxide, the product of iNOS, is known to cause astrocyte swelling.12
Görg et al. identified the cerebral RNA oxidation as a not yet recognized consequence of acute ammonia intoxication. RNA oxidation may affect gene expression and local protein synthesis, and may thereby provide another link between reactive oxygen species/reactive nitrogen oxide species production, and ammonia toxicity. Also, oxidized RNA species may participate in post-synaptic protein synthesis, which is normally a biochemical substrate for learning and memory consolidation.13
Bai et al.14 have proposed that ammonia induces the mitochondrial permeability transition in cultured astrocytes, which may be a factor in the mitochondrial dysfunction associated with HE and other hyperammonemic states (see below).
Glutamate metabolism alterationGlutamate is the main excitatory neurotransmitter in the mammalian CNS. Glutamate modulates several brain processes, including the encoding of information, the formation and retrieval of memories, spatial recognition and the maintenance of consciousness.
Glutamatergic neurotransmission involves several steps, beginning with the release of glutamate from the pre-synaptic neuron; its presence in the extracellular space activates glutamate receptors present in the synaptic membranes, leading to activation of signal transduction pathways related to these receptors. Glutamate is removed from the synaptic cleft by specific glutamate transporters located mainly in astrocytes, avoiding overstimulation of glutamate receptors. The alterations in any of these steps may result in impairment of glutamatergic neurotransmission, leading to neurological alterations, especially those seen in neurodegenerative disease. In addition, the increased excitotoxicity of glutamate produces brain damage, as it also occurs in brain ischemia, anoxia and trauma.
Astrocytic glutamate transportersThere is increasing evidence implying that HE in acute liver failure is the result of altered glutamatergic function. Brain glutamate metabolism constitutes its synthesis, uptake and release, and depends on the last two steps on the normal transporter function; then again, toxic substances and abnormal detoxification mechanisms are responsible for HE syndrome.
Schmidt et al. studied hippocampal slices exposed to serum and cerebrospinal fluid obtained from patients with chronic liver disease who had died in hepatic coma and found that these patients presented an alteration in glutamate uptake that increased glutamate activity.15
Several mechanisms are involved in the uptake of glutamate in astrocytes (Figure 1). In experimental liver damage, after portocaval anastomosis, a decrease in mRNA and protein expression of the glutamate transporter EAAT-2 has been documented16 and similar results have been obtained after ammonia intoxication. In addition, Suarez et al.17 reported that portacaval anastomosis in rats decreases the expression of several glutamate transporters (EAAT-1, EAAT-2, EAAT-3 and EAAC-1). Also, Bender and Noren-berg showed that exposure to ammonia of primary cultures of astrocytes, produces a significant decrease in the high affinity uptake of glutamate.18
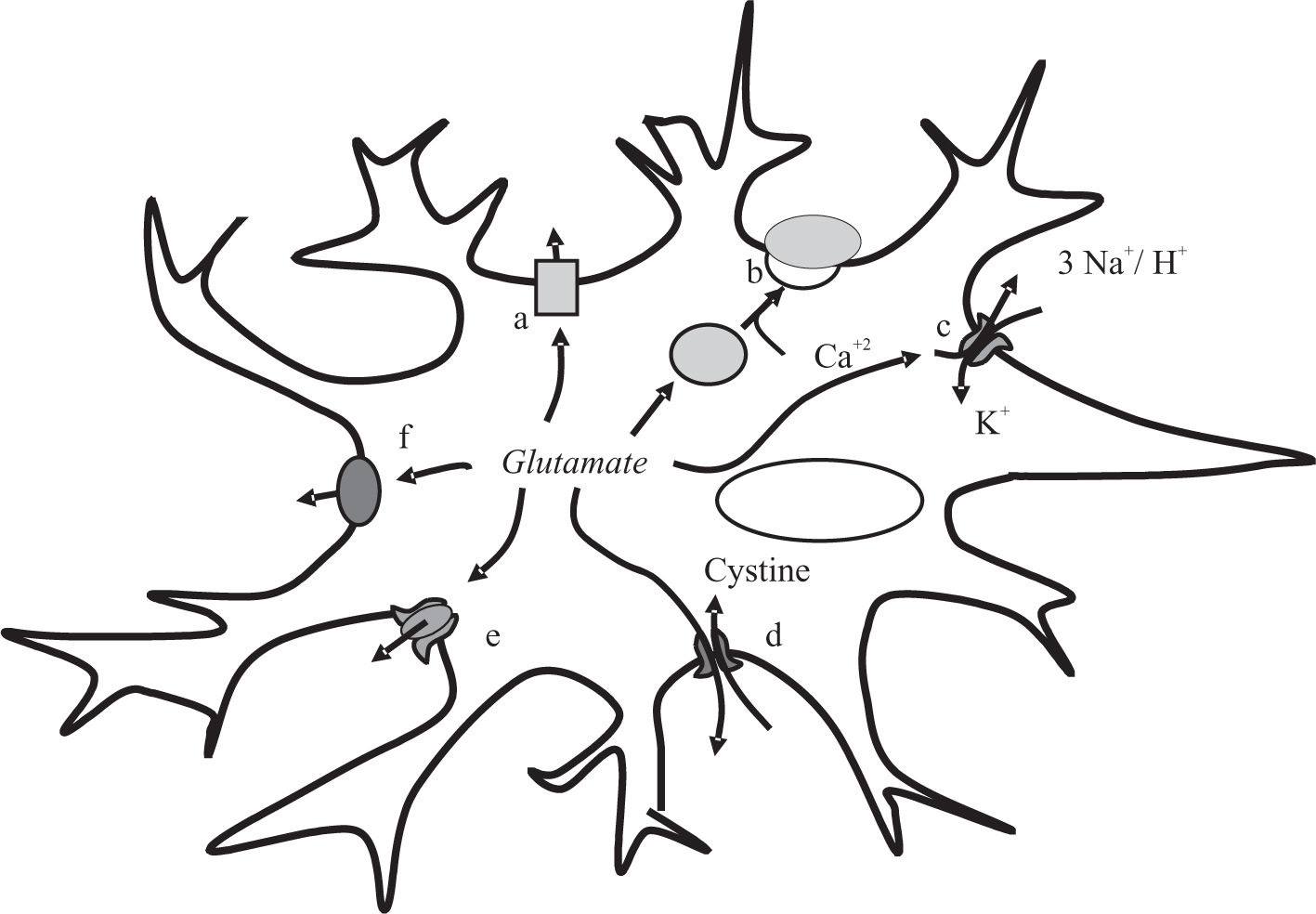
![Glutamate release mechanism in astrocytes. Simplified representation of the six-glutamate release mechanism: aswelling-induced opening of volumeregulated anion channels [VRAC]; bCa2+-dependent exocytosis; creverse operation of excitatory amino acid transporters [EAAT]; d-the cystine-glutamate exchanger; e-connexin hemichannels and f-the P2X7 receptor.](https://static.elsevier.es/multimedia/16652681/0000000800000002/v1_201906260852/S1665268119317855/v1_201906260852/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9DlYm25J0U6w+iS2tsS91ttDPFe/gLIJ6/yM8D23a1sIGu7i2Nr2//2UyxxWTFPWlrbVBMEDucuEQSl7ELPHKZlbOtyCP6yje0QlcLrv4fEmQ46vLQc6zcKL0KOfUFKm20Rlh2YlECiR7wGxymP4peq9iX/Y06n0aN7Rzj148cRqCdFVWr/OQXBoma82azfCnqyh6p3yT06CckKn2d7rNTlIJiZUp/N1mvvRwo8DRiFKgg= "Glutamate release mechanism in astrocytes. Simplified representation of the six-glutamate release mechanism: aswelling-induced opening of volumeregulated anion channels [VRAC]; bCa2+-dependent exocytosis; creverse operation of excitatory amino acid transporters [EAAT]; d-the cystine-glutamate exchanger; e-connexin hemichannels and f-the P2X7 receptor.")
Glutamate release mechanism in astrocytes. Simplified representation of the six-glutamate release mechanism: aswelling-induced opening of volumeregulated anion channels [VRAC]; bCa2+-dependent exocytosis; creverse operation of excitatory amino acid transporters [EAAT]; d-the cystine-glutamate exchanger; e-connexin hemichannels and f-the P2X7 receptor.
Chan et al. demonstrated that the high affinity uptake of glutamate and the astrocytic glutamate transporter GLT-I is decreased in brain when concentrations of ammonia are the same as those reported in brain in acute liver failure. Decreased expression of GLT-1 results in increased extracellular brain glutamate concentrations.19
Also, an increase in extracellular glutamate, as that found in patients with acute liver encephalopathy, emphasizes the importance of ammonia and calcium ions, as well as of pH in the release of glutamate.20
An interesting point found by Tofteng et al. is that patients with intracranial hypertension correlate with high levels of extracellular glutamate in brain.21
Using different models of liver injury, glutamate concentration was found increased in brain tissue and cere-brospinal fluid22,23 in hepatic devascularization24 and in animals exposed to acute hyperammonemia.25
Interestingly, mild hypothermia in rats prevents the initiation of encephalopathy and brain edema, with a decrease in extracellular brain glutamate.26,27
The NMDA receptorGlutamate has two main types of receptors: ionotropic and metabotropic. There are three key types of ionotropic glutamate receptors: NMDA, AMPA and kainate receptor, being the first one implicated in the control of cerebral processes such as neuronal plasticity, learning and memory.28 Metabotropic glutamate receptors are coupled to G proteins and their activation modulates the activity of different enzymes (phospholipase C, adenylate cyclase, etc.) and ion channels through these G proteins, resulting in the modulation of the intracellular levels of second messengers such as diacylglycerol, inositol triphosphates, cAMP, etc. These types of receptors are involved in modulation of motor function, as well.28
Excessive activation of NMDA receptors leads to neuronal degeneration and death. Hyperammonemia can modify the function of NMDA receptors and of some associated signal transduction pathways. The alterations are different in acute and chronic hyperammonemia and liver failure. Acute intoxication leads to an excessive activation of NMDA receptors, which is responsible for ammonia-induced death. In contrast, chronic hyperammonemia induces adaptive responses resulting in impairment of the signal transduction associated with NMDA receptors.29
Activation of NMDA receptors by glutamate leads to increased intracellular Ca2+ in the post-synaptic neurons. Ca2+ binds to calmodulin and activates different enzymes, including nitric oxide synthetase, leading to improved formation of nitric oxide, which activates soluble guanylate cyclase and increases cGMP. According to Felipo et al., this glutamate-nitric oxide-cGMP pathway is impaired in brain in vivo in animal models of HE.28 The impairment of this pathway leads to reduced cGMP levels and contributes to impaired cognitive function in hepatic encephalopathy.
The mitochondrial dysfunction, as a result of an excitotoxic stress, is produced by the over-activation of the NMDA receptor. Calcium homeostasis, ATP production, and formation and detoxification of reactive oxygen species (ROS) participate in this mechanism.30
The glutamate-glutamine cycleThe glutamate-glutamine cycle (Figure 2) is central to our understanding of brain glutamate metabolism and its relation with the brain alteration of HE syndrome. This cycle is fundamental for the detoxification of ammonia.
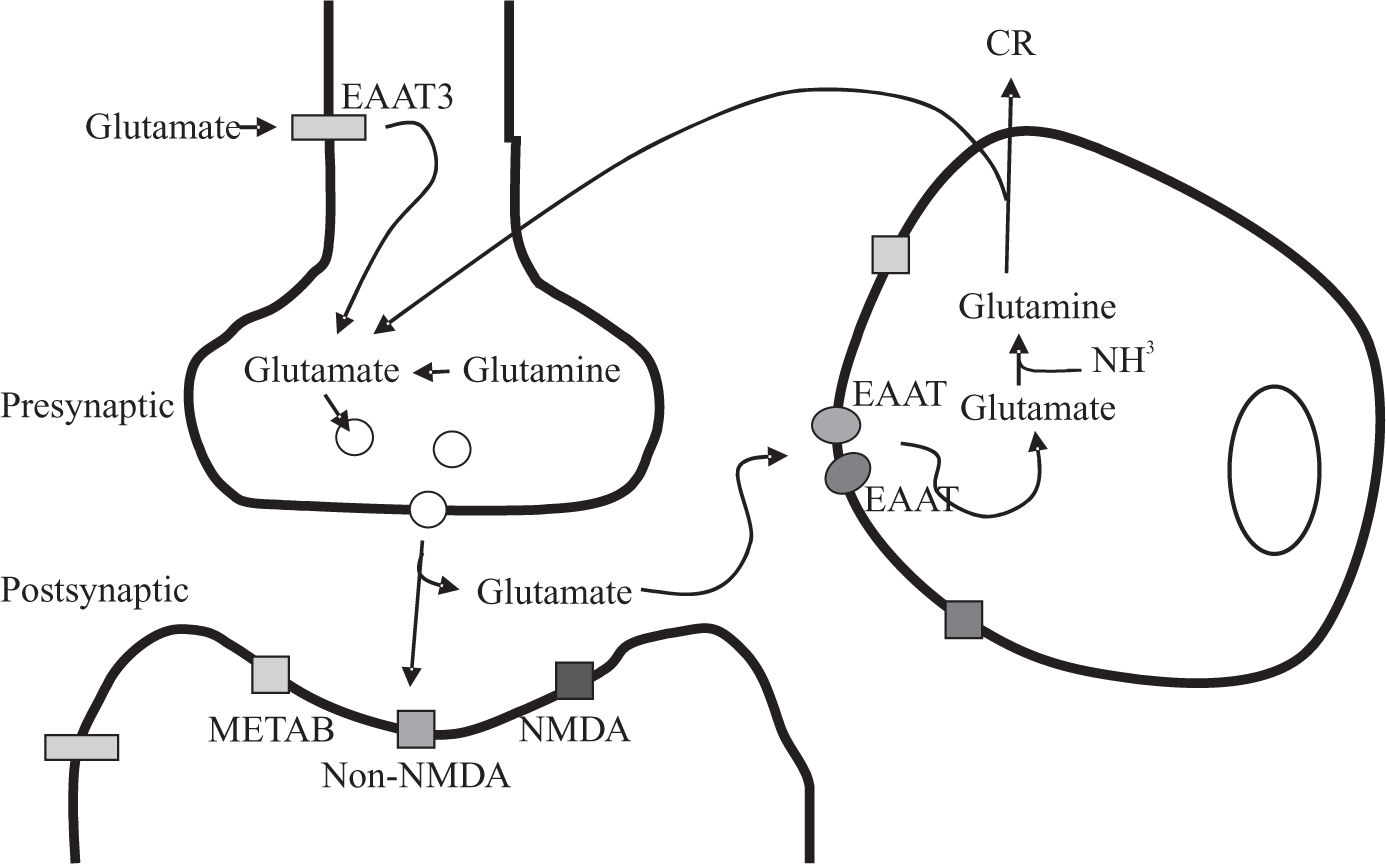
![Schematic representation of the glutamate-glutamine cycle. According to this formulation, glutamate released upon stimulation from presynaptic terminals into the synaptic cleft can activate glutamate receptors [METAB: metabotropic; NMDA and non-NMDA] on post-synaptic neuron or astrocyte. The uptake of glutamate is mediated by the astrocytic glutamate transporters: EAA1 and EAA2. In astrocytes, glutamate is converted to glutamine via the glutamine synthetase pathway. The glutamine is released back to the neurons, where glutamate is regenerated via phosphate-dependent glutaminase, a mitochondrial enzyme.](https://static.elsevier.es/multimedia/16652681/0000000800000002/v1_201906260852/S1665268119317855/v1_201906260852/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9DlYm25J0U6w+iS2tsS91ttDPFe/gLIJ6/yM8D23a1sIGu7i2Nr2//2UyxxWTFPWlrbVBMEDucuEQSl7ELPHKZlbOtyCP6yje0QlcLrv4fEmQ46vLQc6zcKL0KOfUFKm20Rlh2YlECiR7wGxymP4peq9iX/Y06n0aN7Rzj148cRqCdFVWr/OQXBoma82azfCnqyh6p3yT06CckKn2d7rNTlIJiZUp/N1mvvRwo8DRiFKgg= "Schematic representation of the glutamate-glutamine cycle. According to this formulation, glutamate released upon stimulation from presynaptic terminals into the synaptic cleft can activate glutamate receptors [METAB: metabotropic; NMDA and non-NMDA] on post-synaptic neuron or astrocyte. The uptake of glutamate is mediated by the astrocytic glutamate transporters: EAA1 and EAA2. In astrocytes, glutamate is converted to glutamine via the glutamine synthetase pathway. The glutamine is released back to the neurons, where glutamate is regenerated via phosphate-dependent glutaminase, a mitochondrial enzyme.")
Schematic representation of the glutamate-glutamine cycle. According to this formulation, glutamate released upon stimulation from presynaptic terminals into the synaptic cleft can activate glutamate receptors [METAB: metabotropic; NMDA and non-NMDA] on post-synaptic neuron or astrocyte. The uptake of glutamate is mediated by the astrocytic glutamate transporters: EAA1 and EAA2. In astrocytes, glutamate is converted to glutamine via the glutamine synthetase pathway. The glutamine is released back to the neurons, where glutamate is regenerated via phosphate-dependent glutaminase, a mitochondrial enzyme.
The persistence of modified extracellular and intracellular glutamate concentrations also alter the surrounding glial processes, and associated to intrinsic and extrinsic factors may represent the possible first step to apoptosis.
It must always be considered that brain damage associated with liver injury can be associated with apoptotic changes where mitochondria fundamentally participate.31
Another enzyme involved in glutamate metabolism is glutaminase, an enzyme expressed principally by neurons. Glutamate content in cultured cortical neurons diminishes when exposed to ammonia;32 besides, this toxic inhibits phosphate-activated glutaminase.
Ammonia induces depletion of brain glutamate by impairing the malate-aspartate shuttle, which is in charge of the transfer of equivalents between mitochondria and cytosol, ameliorating the reduction of the pyruvate/lactate ratio in cultured astrocytes.33
Morphologically, the blood brain barrier (BBB) is constituted by end feet of astrocytes and endothelial cells with its tight junctions, and apparently is also a target in HE. Glutamate is synthesized in brain from glucose.34 At physiologic plasma concentrations; glutamate flux from plasma into brain is mediated by a high affinity transport system at the BBB. Reverse flux, from brain into plasma, appears to be driven principally by a sodium-dependent active transport system at the capillary abluminal membrane.35 As demonstrated by Scorticati36 and Eizayaga,37 in portal hypertensive rats, BBB increases its permeability. It is thus possible that in this condition glutamate can pass easily from one side to the other of this barrier. Portal hypertension and HE, which are usually present in chronic hepatic disease, allow the easy access of blood vessel contents to neurons and astrocytes.
Glutamine and its relation with brain edemaGlutamine, a neutral amino acid, is normally a non-toxic ammonia carrier in the CNS38,39 and its synthesis represents an astrocyte protection to neurons during increases of blood ammonia concentrations.
The osmotic action of glutamine appears to be responsible for cerebral edema and edema-associated disturbances of cerebral blood flow and ionic homeostasis.38
Vasogenic and cytotoxic mechanisms are implicated in the development of cerebral edema. Vasogenic brain edema occurs as a result of the disruption of the BBB, allowing unrestrained access of plasma components and water to the extracellular cerebral compartment. Cytotoxic edema is the consequence of impaired cellular osmoregulation in the brain, resulting in an increase of cellular water.
There is a good correlation between the increase in glutamine concentration and brain water content. Therefore, an association between ammonia, glutamate and glutamine increases in brain can produce astrocyte swelling in HE, and consequently brain edema.
Astrocytes are the most numerous cell types in the brain and occupy about one-third of the cortical volume. Astrocytes are the cells where glutamine synthesis takes place7 and where water accumulates.40 Then, glutamine-induced astrocyte swelling is one of the fundamental steps in the production of cerebral edema in HE. The impaired cerebral microcirculation can be the result of hyperammonemia, and the addition of the osmotic effect of glutamine.41
After ammonia infusion, methionine-sulfoximine (MSO) treatment, an inhibitor of glutamine synthetase activity, reduces swelling in the rat brain, and prevents the increase in intracranial pressure, when fulminant liver failure or portosystemic anastomosis is present.42 According to Hawkins et al. (1993), in portocaval anastomosed rats pretreated with MSO, an increase in tryptophan uptake is induced, suggesting that the uptake of this amino acid is probably produced by an increase in glutamine content in the brain.43 Actually, the association of ammonia and glutamine seems to be necessary to induce brain edema.
Interestingly, Warren and Shenker observed that the inhibition of glutamine synthetase decreased the death rate of rats intoxicated with ammonia, ameliorating cerebral edema, astrocyte swelling and seizures,44 thus suggesting that some of its bio-products are harmful to brain metabolism.
A good correlation between cerebral glutamine concentration and severity of psychoneurological signs has been found in cirrhotic patients with hyperammonemia;45 besides, glutamine concentration in brain correlates with intracranial pressure measurements.21
According to Albrecht and Norenberg,46 glutamine-mediated ROS production and the mitochondrial permeability transition (MPT) formation are key factors in the production of astrocyte swelling in the presence of ammonia and they proposed the existence of a cascade of events initiated by ROS and MPT.
It is interesting that preventing glutamine degradation in brain mitochondria and glutamine uptake can ameliorate brain damage in HE.46
According to Detry et al., it can be hypothesized that brain edema secondary to the osmotic effect of glutamine in astrocytes, and cerebral hyperemia, secondary to vasodilation (cytokines, products of the necrotic liver, glutamine and others) may contribute to intracranial hypertension leading to brain stem herniation and brain death in fulminant hepatic failure.47
Glutamine synthetaseGlutamine synthetase (GS) is a key enzyme in glutamate metabolism (Figure 2), and its decreased activity leads to glutamate increases in the extracellular space. Then, the synthesis of glutamine from glutamate and ammonia in the brain is an important step for detoxification of ammonia in hepatic failure with hyperammonemia.48
Glutamine synthesis takes place in astrocytes and maintains glutamate homeostasis in the brain, avoiding an excess of this amino acid, and thus an excitotoxic condition. Glutamine has a short half-life and its activity is regulated and modulated by several mediators and hormones.
Nevertheless, as it occurs in severe liver insufficiency, brain astrocytes are injured, morphologically and functionally, and actually GS activity must be involved in this damage.
Derouiche and Frotscher have suggested that extracellular glutamate can regulate GS distribution, and that glutamate influences the perisynaptic astrocytic processes with the production of damage in glutamatergic zones.49
A correct functioning of glutamine synthetase in astrocytes leads to a decrease in hyperammonia, and consequently, to a decrease in brain glutamate concentration; in contrast an altered functioning of this pathway allows glutamate to increase.50
Oxidative stress in brainReactive oxygen species have been implicated in the pathophysiology of acute and chronic liver disease.
The existence of an oxidative stress (OS) interferes with many nervous cell functions, creating an up-regulation of cytokines and adhesion molecules, and inducing the presence of pro-apoptotic proteins.
Murthy et al.51 and Song et al.52 demonstrated the presence of OS in brain tissue in the presence of liver failure. Blanc et al. observed that in liver damage, the activity of glutamate transporters is also impaired.53
Mitochondrial dysfunction and morphological damage is present, and MPT can be formed with the consequent extrusion of its matrix content.54
OS is responsible, then, for the MPT production in mitochondria, and lipid and protein peroxidation in its membranes.
According to Tirosh et al.,55 HT4 cells obtained from mouse hippocampal brain region, exposed to glutamate for twelve hours, produce a rapid loss of intracellular reduced glutathione. This is followed by an intracellular accumulation of ROS, increases in intracellular Ca2+, loss of mitochondrial membrane potential and swelling.30
TNF•, ceramide and bile acids enhance ROS production, as well.
OS depletes ATP by inhibiting glycogenesis, thus producing an important energy deficiency. The result of the presence of ROS depends on the number of cells involved and the functioning of the anti-stress systems, an adequate glutathione system, a correct antitoxic mechanism, and the activities of antioxidant enzymes such as catalase, superoxide dismutase and glutathione peroxidase.
OS participates as a critical factor in the mechanism of HE production, especially in hyperammonemic conditions.56,57
Increased brain production of nitric oxide has been found in rat portocaval anastomosis, which can also participate in the formation of OS.
Recently, in a clinical study in which we studied 30 cirrhotic patients, we showed that among those with liver decompensation, OS parameters demonstrated a significant increase, as shown by the presence of the thiobarbituric acid reactive substances, and a decrease in catalase and glutathione peroxidase activity, from the antioxidant enzymes system.58
Apparently, there is no clear evidence for the loss of high-energy phosphates in liver damage, although there are defects in the tricarboxylic acid cycle.59
Manganese accumulation in brain structures has also been implicated in liver failure brain damage.60,61
ConclusionThe metabolic alterations that appear in HE can be reversible if the brain damage is not extensive and if the time of toxins action is not prolonged.
In agreement with Abou-Assi and Vlahcevic, we think that brains of patients with chronic liver disease present impairments in more than a few metabolic pathways, a fact that could explain, in part, the neuropsychiatric signs found in HE.
Finally, taking all these experimental and clinical data together, it can be suggested that ammonia is not the only toxic agent that induces HE, but several other substances participate in the development of the encephalopathic syndrome in liver acute and chronic damage, namely the increase in the excitotoxic glutamate levels, the increased concentration of glutamine and the emergence of ROS. It must be recalled that the presence of oxidative stress in brain without adequate defenses aggravates the CNS condition.



