



Background. Diagnosis of progressive familial intrahepatic cholestasis (PFIC) is a challenging matter that involves the summation of clinical, laboratory, radiological, and liver histological parameters; in addition to specific investigations to exclude other causes of neonatal cholestasis. The aim of this study was to evaluate liver tissue immunohistochemistry of bile salt export pump (BSEP) and multidrug resistance 3 (MDR3) proteins in differentiating PFIC from other causes of neonatal cholestasis, particularly, when genotyping is unavailable.
Material and methods. The study included 25 patients diagnosed phenotypically as PFIC including 2 with PFIC1, 17 with PFIC2 and 6 with PFIC3. A second group of 25 cholestatic newborns with confirmed etiologies other than PFIC, termed as non-PFIC, included as controls. Liver biopsies from all patients were obtained and immunostained for BSEP and MDR3.
Results. Negative immunoreaction of BSEP and MDR3 was found in the majority of PFIC group (76 and 64% respectively). Nonetheless, the negative immunoreaction was demonstrated in a considerable number of the non-PFIC group. BSEP immunoreaction was negative in the majority (82.4%) of PFIC2 but in none of the two patients with PFIC1. In addition, negative MDR3 immunoreaction was more frequently associated with PFIC3 compared to non-PFIC group.
Conclusion. MDR3 and BSEP immunostaining would be a helpful tool in supporting the phenotypic diagnosis of PFIC subtypes and in differentiating PFIC from other causes of neonatal cholestasis.
Neonatal cholestasis constitutes a serious medical condition being the main manifestation of hepatobiliary disease and having a wide spectrum of causes that arise from abnormalities in the uptake, handling, transport, and excretion of bile salts and bilirubin by hepatocytes or in the flow of bile through the bile canaliculi and ducts.1
Progressive familial intrahepatic cholestasis (PFIC) refers to a heterogeneous group of autosomal-recessive disorders that disrupt bile formation and present with cholestasis of hepatocellular origin. Three types of PFIC have been identified. PFIC1 and PFIC2 usually appear in the first months of life, whereas the onset of PFIC3 may arise later in infancy, in childhood or even during young adulthood. The main clinical manifestations include pruritus and jaundice. PFIC patients usually develop fibrosis and end-stage liver disease before adulthood.2
Both PFIC1 and PFIC2 are caused by impaired bile salt secretion due to defects in ATP8B1 (adenosine triphosphate, type 8B, member 1) gene encoding the FIC1 protein and in ABCB11 (adenosine triphosphate-binding cassette, subfamily B, member 11) gene encoding bile salt export pump (BSEP) protein, respectively. Defects in ABCB4 (ABCB member 4) gene, encoding multidrug resistance 3 protein (MDR3), impair biliary phospholipid secretion, resulting in PFIC3.3 ATP8B1 is abundantly expressed in a wide variety of tissues such as the small intestine, bladder and stomach and to a lesser extent also in the liver and pancreas. This results in the multitude of the extrahepatic manifestations such as hearing loss, pancreatitis and diarrhea, found in patients with PFIC1.4
PFIC should be suspected in children with a clinical history of cholestasis of unknown origin after exclusion of other common causes of cholestasis (such as biliary atresia [BA], alagille syndrome, alpha-1-antitrypsin deficiency, cystic fibrosis, sclerosing cholangitis and extrahepatic bile duct obstruction). Generally, patients with PFIC1 and PFIC2 have normal serum gamma glutamyl transpeptidase (GGT), while patients with PFIC3 have high GGT.5 Ultimately, the diagnosis of PFIC remains a challenging matter. MDR3 and BSEP liver immunostaining, although inconclusive in many cases, help to select PFIC candidates in whom molecular diagnosis can be proposed.6–8
We aimed to evaluate of the role of BSEP and MDR3 immunohistochemistry in differentiating PFIC from other causes of neonatal cholestasis and in allocating the different PFIC types.
Material and MethodsStudy populationThis retrospective study included 50 infants with neonatal cholestasis (NC). Patients were recruited from the Pediatric Hepatology department, National Liver Institute, Menoufiya University. According to the final diagnosis, patients were divided into two groups: a PFIC group (n = 25) included infants diagnosed phenotypically (nongenetic) as PFIC. A non-PFIC group (n = 25) of cholestatic newborns with confirmed diagnoses other than PFIC served as controls. The study was approved by the Ethics Review Board of the National Liver Institute.
Etiological diagnosisAfter complete history taking, thorough clinical examination, routine investigations, (liver function tests, prothrombine time, complete blood count, hepatitis viral markers, TORCH markers) and total serum bile acid measurement, phenotypic diagnosis of PFCI was based on a constellation of a clinical, biochemical and histopathological studies together with the exclusion of other causes of neonatal cholestasis. These criteria included low or normal GGT in PFIC1 and PFIC2 and high GGT in PFIC3, elevated serum transaminases, elevated serum total bile acids, ultrasonography (US) of the liver revealing early hepatomegaly and excluding biliary tract disease. Liver biopsy examination showed the characteristics of each type of PFIC. Patients with PFIC1 had bland cholestasis (with almost no inflammation), fibrosis, ductular proliferation and canalicular bile plugs. In PFIC2, they had giant cell hepatitis, fibrosis and duct reaction. In PFIC3, they had ductular proliferation and mixed inflammatory infiltrates and cholestasis with slight giant cell transformation. Older patients had extensive portal fibrosis. Pruritus was evident in some patients. PFIC1 was distinguished from PFIC2 by the presence of extrahaptic manifestations in PFIC1 in the form of persistent diarrhea. GGT levels were considered according to the expected for the age.9 The diagnoses of cholestasis group were determined by specific laboratory tests according to the expected etiology, liver biopsy, operative cholangiography prior to Kasai portoenterostomy in BA patients. The diagnoses in this group were BA (n = 14), galactosemia (n = 3), paucity of intrahepatic bile ducts (n = 2), bile acid synthetic disorder (n = 2), tyrosinemia (n = 1), ARC (arthrogryposis, renal dysfunction and cholestasis) syndrome (n = 1), choledochal cyst (n = 1) and Edward’s (Trisomy 18) syndrome (n = 1). The severity of liver disease was assessed by the pediatric end-stage liver disease (PELD) score.10
Liver biopsy and BSEP and MDR3 immunohistochemistryUltrasonography-guided liver biopsy was performed for all patients using Tru-cut needle. Biopsy specimens were fixed in formalin-buffered saline, embedded in paraffin and stained by hematoxylin and eosin, Masson’s trichrome, Orcein and Perl’s stains for routine histopathological evaluation. Portal fibrosis was scored according to Ishak scoring system.11 Immunohistochemical staining of BSEP and MDR3 was performed as described in Evason. et al.8 Briefly, three 4-μm-thick sections from each paraffin block were cut, mounted on Superfrost Plus slides (Thermo Fisher Scientific Inc., Waltham, MA, USA), deparaffinized in xylene and rehydrated in descending grades of ethyl alcohol. Endogenous peroxidase was blocked using 0.3% H2O2 in phosphate-buffered saline (PBS). After two washes with PBS, sections were incubated with biotin blocking system (Dako North America Inc. Carpintería, CA 93013, USA) followed by incubation with antigen retrieval solution (Dako North America Inc. Carpinteria, CA 93013, USA) for 10 min then 5 washes with distilled water. Slides were incubated with primary antibody using mouse ABCB11/BSEP anti-human monoclonal antibody, and rabbit ABCB4/MDR3 anti-human polyclonal (N-terminus) antibody (both were provided by LifeSpan BioSciences, Inc. (LSBio), Seattle, WA, USA. Catalogue No. LS-B2170 and LS-B5729 respectively). Normal liver and placental tissues were included as positive controls for BSEP and MDR3 staining respectively. After several washes in PBS, the slides were incubated with biotin-conjugated secondary antibody using goat antimouse anti-rabbit immunoglobulins and then incubated with streptavidin-HRP (horseradish peroxidase). The reaction was visualized using diaminobenzidine followed by Hematoxylin counterstaining (All were done using LSAB2 System-HRP, Dako North America Inc. Carpinteria, Ca 93013, USA). Immunostained sections were evaluated by a liver histopathologist who was unaware of the diagnosis of the patients. The expression of positive cells was interpreted in a qualitative way based on the presence or absence of the immunoreaction and was expressed as either positive or negative.
Statistical analysisDescriptive results were expressed as mean ± standard deviation (SD) or number (percentage) of individuals with a condition. Statistical significance between groups was tested by the nonparametric Mann-Whitney U test for quantitative data, and Pearson’s χ2 test or Fisher’s exact test for qualitative data. Significance among more than two groups was tested by Kruskal-Wallis test. The diagnostic performance was measured as sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) and all were expressed as percentage. Results were considered significant if p ≤ 0.05. Statistical analysis was performed using SPSS software version 13 (SPSS Inc, Chicago, IL, USA).
ResultsStudy population’s characteristicsThe current study included 50 infants divided into PFIC group (n = 25) and non-PFIC group (n = 25). Both were age (208.68 ± 229.69 vs. 104.96 ± 147.15 respectively) and sex (M%/F%; 64/36 vs. 52/48 respectively) matched (P > 0.05 for both). The age in PFIC group ranged from 42 to 912 days with a median of 113 days. The age in non-PFIC group ranged from 23 to 730 days with a median of 60 days. The frequency of pruritis was significantly higher in PFIC1 and PFIC2 compared to non-PFIC group while the frequency of clay-colored stool and abnormal gallbladder were significantly higher in the non-PFIC group when compared to each PFIC group. Transaminases levels in PFIC1 and GGT levels in PFIC1 and PFIC2 were significantly lower than the levels in non-PFIC group. The mean values of serum bile acids were significantly higher in PFIC1 and PFIC2 groups than in the non-PFIC group. PELD score in PFIC3 was significantly lower than in non-PFIC group while other parameters were comparable between each PFIC group and the non-PFIC group (Table 1).
Clinical, ultrasonographic and laboratory characterstics of the study population.
| Parameter | PFIC-1 (n = 2) | PFIC-2 (n = 17) | PFIC-3 (n = 6) | Non-PFIC (n = 25) |
|---|---|---|---|---|
| Pruritus | 1 (50)* | 6 (35.3)* | 0 (0.0) | 0 (0.0) |
| Clay stool | 0 (0.0)* | 3 (17.6)* | 1 (16.7)* | 14 (56) |
| Hepatomegaly (US) | 2 (100) | 13 (76.5) | 6 (100) | 14 (56) |
| Splenomegaly (US) | 2 (0.0) | 9 (52.9) | 4 (66.7) | 11 (44) |
| Abnormal gallbladder (non-contractile or atretic) | 0 (0.0)* | 3 (17.6)* | 2 (33.3)* | 19 (76) |
| Total bilirubin (mg/dL) | 11.75 ± 3.89 | 11.41 ± 5.84 | 7.95 ± 2.79 | 12.76 ± 5.24 |
| Direct bilirubin (mg/dL) | 9.05 ± 2.62 | 7.82 ± 4.37 | 6.08 ± 2.11 | 8.96 ± 3.58 |
| Albumin (g/dL) | 3.55 ± 0.35 | 3.29 ± 0.56 | 3.45 ± 0.26 | 3.12 ± 0.56 |
| Aspartate transaminase (U/l) | 53 ± 8.48* | 437.24 ± 682.22 | 231.17 ± 107.67 | 306.16 ± 222.17 |
| Alanine transaminase (U/l) | 24.5 ± 2.12* | 225.71 ± 296.14 | 118.83 ± 45.45 | 222.72 ± 223.89 |
| Gamma glutamyle transpeptidase (U/l) | 33.5 ± 20.51* | 49.94 ± 27.12* | 361 ± 230.28 | 665.96 ± 677.89 |
| Alkaline phosphatase (U/l) | 427.5 ± 99.7 | 560.88 ± 371.46 | 466.67 ± 191.62 | 456.44 ± 266.63 |
| INR | 1.2 ± 0.28 | 1.29 ± 0.8 | 1.02 ± 0.04 | 1.06 ± 0.09 |
| Serum bile acids (μmol/l) | 171 ± 41.01* | 145.65 ± 104.55* | 106.98 ± 92.26 | 30.87 ± 52.08 |
| PELD score | 16.5 ± 0.71 | 11.24 ± 6.78 | 6.5 ± 6.4* | 10.56 ± 4.54 |
INR: international normalized ratio. US: ultrasonography. PELD: pediatric end-stage liver disease.
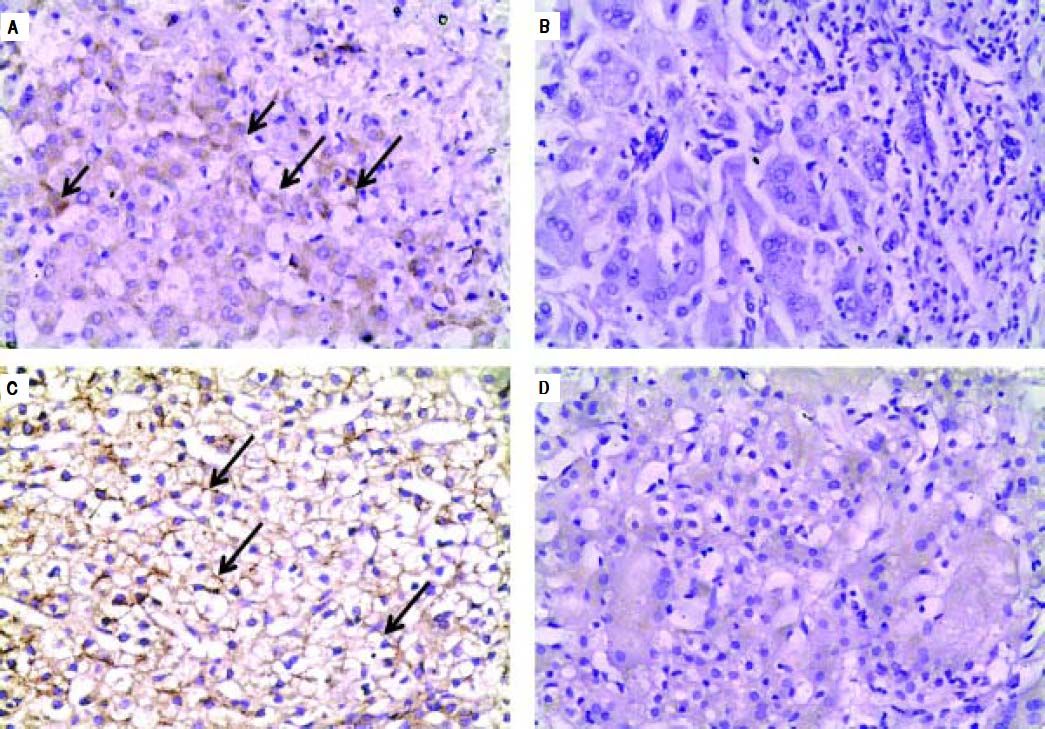
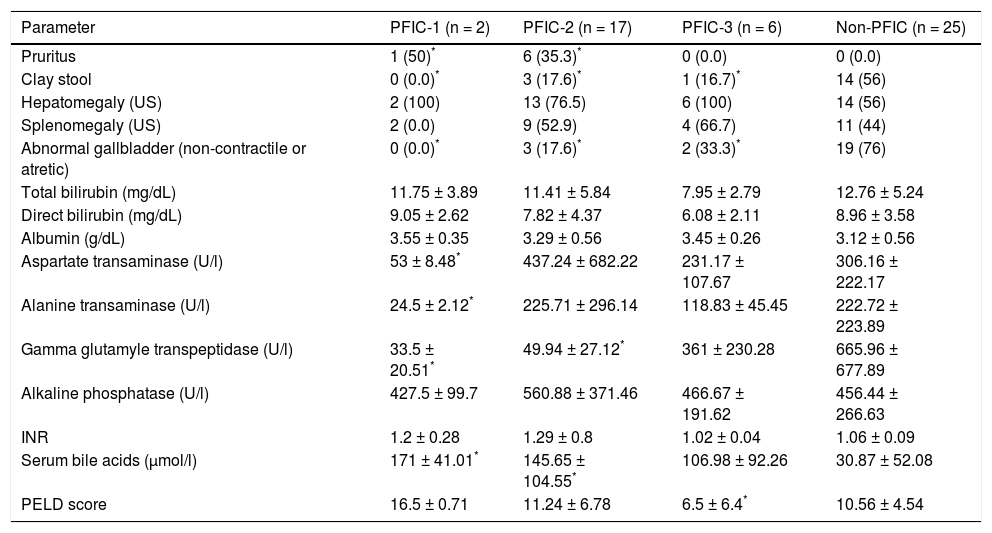
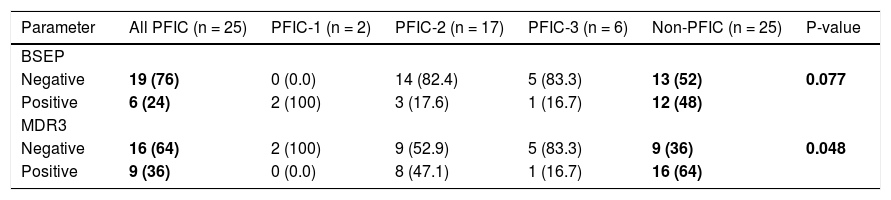
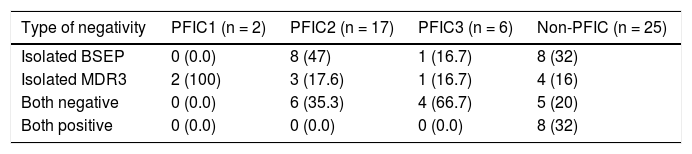
Negative IHC staining of BSEP and MDR3 was found in the majority of PFIC group (76 and 64% respectively). Nonetheless, the negative expression was demonstrated in a considerable number of patients in the non-PFIC group (52 and 36% for BSEP and MDR3 respectively) with significant statistical difference for MDR3 (P = 0.048) but not for BSEP (P = 0.077) (Table 2). When comparing the expression in the PFIC subtypes, BSEP was negative in the majority of PFIC type 2 and 3 but in none of the two patients with PFIC type 1. Similarly, negative MDR3 immunostaining was found in the majority of all PFIC subtypes, while it was frequently associated with PFIC type 3 (83.3%) compared to non-PFIC group (36%) (Table 2). The number of patients in each PFIC subgroup was small to calculate a statistical significance. Positive immunostaining of BSEP was noticed in the canalicular membrane and the cytoplasm of hepatocytes and that of MDR3 was noticed in the canalicular membrane (Figure 1).
BSEP and MDR3 immunostaining in the studied groups.
| Parameter | All PFIC (n = 25) | PFIC-1 (n = 2) | PFIC-2 (n = 17) | PFIC-3 (n = 6) | Non-PFIC (n = 25) | P-value |
|---|---|---|---|---|---|---|
| BSEP | ||||||
| Negative | 19 (76) | 0 (0.0) | 14 (82.4) | 5 (83.3) | 13 (52) | 0.077 |
| Positive | 6 (24) | 2 (100) | 3 (17.6) | 1 (16.7) | 12 (48) | |
| MDR3 | ||||||
| Negative | 16 (64) | 2 (100) | 9 (52.9) | 5 (83.3) | 9 (36) | 0.048 |
| Positive | 9 (36) | 0 (0.0) | 8 (47.1) | 1 (16.7) | 16 (64) |
P-value represents all PFIC vs. non-PFIC
 and cytoplasm of hepatocytes (short arrow). B. Negative immunoreaction of BSEFin liver tissue from a case of PFIC2. C. Immunostaining of MDR3 in liver tissue from a case of non-PFIC. Positive immunostaining was noticed in canalicular membrane (long arrow). D. Negative immunoreaction of MDR3 in liver tissue from a case of PFIC3 (original mag. x 200).")
Immunohistochemical staining of BSEP and MDR3. A. Immunostaining of BSEP in liver tissue from a case of non-PFIC. Positive immunostaining was noticed in canalicular membrane (long arrow) and cytoplasm of hepatocytes (short arrow). B. Negative immunoreaction of BSEFin liver tissue from a case of PFIC2. C. Immunostaining of MDR3 in liver tissue from a case of non-PFIC. Positive immunostaining was noticed in canalicular membrane (long arrow). D. Negative immunoreaction of MDR3 in liver tissue from a case of PFIC3 (original mag. x 200).
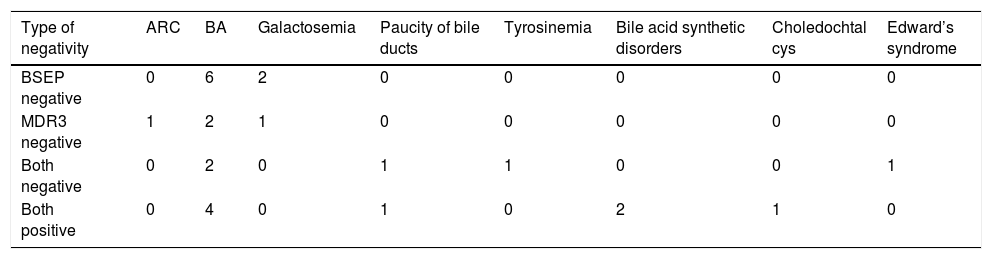
The negative immunoreaction of BSEP and MDR3 was found either isolated or in combination. None of the PFIC group was positive for the two stains simultaneously. Combined negativity was more frequent (66.7%) in PFIC3 while isolated BSEP negativity was more frequent (47%) in PFIC2 (Table 3). The type of BESP and/or MDR3 negativity in different non-PFIC diagnoses was as demonstrated in table 4.
Type of IHC negativity in PFIC1, PFIC2, PFIC3 and non-PFIC cholestasis groups.
| Type of negativity | PFIC1 (n = 2) | PFIC2 (n = 17) | PFIC3 (n = 6) | Non-PFIC (n = 25) |
|---|---|---|---|---|
| Isolated BSEP | 0 (0.0) | 8 (47) | 1 (16.7) | 8 (32) |
| Isolated MDR3 | 2 (100) | 3 (17.6) | 1 (16.7) | 4 (16) |
| Both negative | 0 (0.0) | 6 (35.3) | 4 (66.7) | 5 (20) |
| Both positive | 0 (0.0) | 0 (0.0) | 0 (0.0) | 8 (32) |
Type of negativity in different non-PFIC diagnoses.
| Type of negativity | ARC | BA | Galactosemia | Paucity of bile ducts | Tyrosinemia | Bile acid synthetic disorders | Choledochtal cys | Edward’s syndrome |
|---|---|---|---|---|---|---|---|---|
| BSEP negative | 0 | 6 | 2 | 0 | 0 | 0 | 0 | 0 |
| MDR3 negative | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 0 |
| Both negative | 0 | 2 | 0 | 1 | 1 | 0 | 0 | 1 |
| Both positive | 0 | 4 | 0 | 1 | 0 | 2 | 1 | 0 |
PFIC represents 10 to 15% of causes of cholestasis in infants and a comparable percentage of the indications for liver transplantation in the pediatric age group.5 Diagnosis of PFIC may be challenging. Identification of a genetic mutation is helpful, but this analysis is not yet readily available in clinical practice in Egypt.1 The aim of our study was to evaluate the diagnostic utility of BSEP and MDR3 immunostaining in differentiating PFIC from other causes of neonatal cholestasis and differentiating subtypes of PFIC from each other.
In the current study, negative BSEP immunostaining in liver tissues showed no significant difference between PFIC (76%) and non-PFIC (52%) groups (P = 0.077). On the other hand, when comparing the low GGT types of PFIC, BSEP negative immunostaining was frequently associated with PFIC2 (82.4%) compared to none of the two patients with PFIC1, but the small number of patients in PFIC1 was not sufficient to evaluated the clinical significance.
The frequency of negative BSEP immunostaining in PFIC2 patients is consistent with that of Evason, et al.,8 who reported 83.3% negative BSEP expression in PFIC2 patients with positive homozygous or heterozygous ABCB11 mutation. Similarly, Davit-Spraul, et al.6 revealed a negative BSEP expression in 91.7% of PFIC2 patients with positive homozygous or heterozygous ABCB11 mutation. The positivity of BSEP expression, in spite of the positive homozygous or heterozygous ABCB11 mutations in the latter two studies, can be explained by the fact that some ABCB11 mutations with disease-causing effects are likely to result in abnormal location or function of the protein but with retained antigenicity.12
Our results in the low-GGT PFIC types (PFIC1 and PFIC2) along with the previous studies that compare IHC results to genotyping suggests that a small proportion of low-GGT PFIC is likely not due to ATP8B1 or ABCB11 mutations and the possible existence of other disease loci.6,13,14
We found that negative MDR3 immunostaining was significantly higher in PFIC group (64%) compared to non-PFIC (36%) group (P = 0.048). In contrast, negative MDR3 immunostaining was comparable among PFIC3 (83.3%), PFIC1 (100%) and PFIC2 (52.9%). Colombo, et al.7reported absent or faint MDR3 staining in 71.42% of PFIC3 cases of whom only 80% had homozygous ABCB4 mutations and the remaining 20% had heterozygous mutations. He suggested that, regardless the type of ABCB4 gene mutations, the simple heterozygous state is associated with a highly variable phenotypic spectrum from complete absence of symptoms during childhood to a clinical history characterized by transient neonatal cholestasis followed by contraceptive-induced cholestasis and the presence of compensated cirrhosis with evidence of portal hypertension by late teens. In the same context, Wendeum, et al.15 reported that in PFIC3 patients who had ABCB4 gene mutation, MDR3 immunostaining can show a complete absence of canalicular staining, a faint or sometimes normal canalicular staining.
PFIC3, being the only type of PFIC with high-GGT, loses the advantage of being discriminated from other common causes of cholestasis by GGT level, since most of the latter present with high GGT. On that basis, we compared negative MDR3 immunostaining in PFIC3 group with that of non-PFIC group to establish grounds for differentiation between both groups. MDR3 negativity was frequently associated with PFIC3 patients (83.3%) compared to non-PFIC patients (36%). Thus, in someway, it may support the discrimination of PFIC3 from non-PFIC patients.
Of interest, BSEP and MDR3 negativity were not restricted to PFIC patients. We found isolated negativity of BSEP in two (8%) patients of the non-PFIC group. One had BA and the other had galactosemia. Chen, et al.16 found that in early-stage obstructive cholestasis (such as BA), the liver showed rapid adaptive down-regulation in BSEP and MDR3. However, the isolated negativity of BSEP requires a further study. Furthermore, Liu, et al.17 reported that PFIC genes’ mutations were found in neonatal cholestasis patients with defined etiologies other than PFIC. For that, genetic profiling in such patients, as those with galactosemia, for ABCB11 mutations need to be considered.
In our study, isolated negativity of MDR3 was found in three (17.6%) patients of PFIC2 group and in four (16%) patients of the non-PFIC group. The latter were two with BA, one with ARC syndrome and one with galactosemia. Negative MDR3 immunostaining in the low-GGT PFIC2 raises the point of ABCB4 mutations presenting with low-GGT cholestasis. Schneider, et al.18 reported a case of intrahepatic cholestasis of pregnancy (ICP) with ABCB4 mutation in whom, GGT activity during ICP was normal in two pregnancies. Although it has been reported that GGT is a reliable marker to differentiate between PFIC1 and BSEP defects at one end and MDR3 defects at the other one, the low-GGT values found in that ICP patient support the view that low-GGT values do not exclude ICP linked to MDR3 mutations. This finding may apply to other forms of ABCB4 mutation-related diseases such as PFIC3. Further substantiation of this hypothesis requires genotyping of these cases.
In the current study, two of the three PFIC2 patients with isolated MDR3 negativity had markedly elevated serum transaminases (2,876 and 1,226 U/l in the first and 913 and 541 U/l in the second for AST and ALT respectively), with otherwise laboratory and histopathological findings supporting the diagnosis of PFIC2. The only parameter that advocates PFIC3 in the two cases is the markedly elevated transaminases. However, phenotypic diagnosis of PFIC3 in these two cases was strongly contradicted by the low GGT levels in both.
MDR3 negativity in BA patients draws the attention to the findings of Menchise. et al.,19 who reported an infant with BA with the anatomical and molecular features of ABCB4/MDR3 deficiency. In his study of a murine model, ABCB4 heterozygosity enhanced hepatic CD8 and natural killer lymphocytes responses to virus infection and increased the frequency of ductal obstruction. The negative immunoreaction of MDR3 in ARC and galactosemia patients requires further investigations and genotyping since there is no relevant studies or literature on the subject.
We found a combined BSEP and MDR3 negativity in 35.3% of PFIC2 group, 66.7% of PFIC3 group and in 20% of the non-PFIC group. Those in the non-PFIC group were two BA patients and a case of Edward’s syndrome. Combined BSEP and MDR3 negativity could be attributed, in part; to the possible existence of combined homozygous mutations of ABCB11 and ABCB4 genes as reported by Keitel, et al.,20 in a patient with ICP. In addition, prior case reports linked Edward’s trisomy to BA21,22 and another case report linked trisomy 18 to paucity of bile ducts.23 In BA, the down-regulation of canalicular and sinusoidal uptake transporters mentioned previously could explain the absence of both BSEP and MDR3 proteins in liver biopsies of such patients.16 In paucity of bile ducts, the hepatocytes are directly exposed to the detergent effect of bile acids by the lacking of the bile ducts in portal tracts which results ultimately in biliary cirrhosis.24
The diagnosis of PFIC is tentative and dependent on clinical parameters (i.e., phenotype, serum GGT and bile acid level), combined with liver biopsy findings.25 Genotyping, when available, helps to confirm the diagnosis. Nonetheless, the genotype-phenotype correlation is not always clear indicating the probable involvement of additional genes in PFIC. A better understanding of the phenotype-genotype correlation in PFIC will lead to improved diagnoses and treatments.26 For that, the inability to perform genotyping is a limitation in our study.
ConclusionIn conclusion, the current study demonstrated that, BSEP staining was not able to differentiate between the PFIC cohort and non-PFIC patients. Only MDR3 negativity discriminated between PFIC3 and non-PFIC patients, however the small number of patients in the PFIC subtypes (PFIC1 = 2 and PFIC3 = 6) is another limitation in the current study. Nonetheless, our results make a further study on a larger PFIC cohort worthwhile. Furthermore, the negative immunostaining in the non-PFIC group opened horizons for the possible molecular events similar to that of PFIC occurring in non-PFIC patients. Whether these events occurred as a consequence of cholestasis or have a genetic basis can only be answered with a thorough genetic profiling of PFIC genes in such patients.
Abbreviations- •
ARC: arthrogryposis, renal dysfunction and cholestasis.
- •
ATP8B1: adenosine triphosphate, type 8B, member 1.
- •
BA: biliary atresia.
- •
BSEP: bile salt export pump.
- •
GGT: gamma glutamyl transpeptidase.
- •
IHC: immunohistochemistry.
- •
MDR3: multidrug resistance 3.
- •
NC: neonatal cholestasis.
- •
NPV: negative predictive value.
- •
PELD: pediatric end-stage liver disease.
- •
PFIC: progressive familial intrahepatic cholestasis.
- •
PPV: positive predictive value.
This study was funded by the National Liver Institute, Menofiya University, Egypt, without any particular role in the study design, data collection and analysis or the writing of the report.
Conflict Of InterestThe authors declare that they have no competing interest.



