



Type 2 diabetes mellitus (T2DM) increases the occurrence and mortality of liver cancer. Insulin growth factor (IGF) plays a crucial role in the development of diabetes and liver cancer, and specifically, IGF-1 may be involved in the development of liver cancer with preexisting T2DM. Autophagy contributes to cancer cell survival and apoptosis. However, the relationship between IGF-1 and autophagy has rarely been evaluated. The purpose of this study was to investigate whether IGF-1 promotes the development of liver cancer in T2DM patients by promoting autophagy.
Materials and methodsThirty-three hepatocellular carcinoma (HCC) patients with T2DM and 33 age-matched patients with HCC without T2DM were included in this study. We analyzed the expression of IGF-1 and autophagy-related LC3 and p62 mRNA and the prognosis of two groups. In vitro, we stimulated HepG2 cells with IGF-1 and then detected changes in autophagy and cell proliferation, apoptosis, and migration in the presence/absence of wortmannin, an autophagy inhibitor.
ResultsIGF-1 promoted autophagy, resulting in inhibition of apoptosis and induction of growth and migration of HepG2 cells. Inhibition of autophagy by wortmannin impaired IGF-1 function. Higher expression of IGF-1 was detected in HCC patients with T2DM. IGF-1 expression was higher in liver cancer tissue compared to paracancerous tissue. Elevated IGF-1 was associated with a poor prognosis in patients with HCC.
ConclusionsIGF-1 participates in the development of liver cancer by inducing autophagy. Elevated IGF-1 was a prognostic factor for patients with HCC, especially when accompanied by T2DM.
Liver cancer is one of the most common and fatal malignancies, and the treatment options are extremely limited. Approximately 75%–85% of primary liver cancers are hepatocellular carcinoma (HCC) [1]. There are many risk factors for liver cancer, including viral hepatitis, metabolic liver disease, alcohol addiction, and exposure to dietary toxins [1]. Diabetes mellitus (DM) is a metabolic disorder characterized by hyperglycemia, which can predispose the liver to insulin resistance and induce liver injury, both due to inadequate insulin secretion or insulin receptor insensitivity to endogenous insulin [2]. Type 2 Diabetes Mellitus (T2DM) has been reported to cause an approximately 2-fold to 3-fold increased risk of liver cancer and increase the morbidity and mortality of liver cancer when compared to non-diabetic subjects [3, 4]. Insulin resistance, reactive oxygen species, hyperinsulinemia, and inflammation are believed to promote hepatocarcinogenesis. Aberrations in insulin-like growth factor-1 (IGF-1) signaling are also a potential mechanism of diabetes-related hepatocarcinogenesis [5].
IGF-1 is an endocrine and autocrine/paracrine growth factor, and approximately 75% of IGF-1 bound forms are synthesized by the liver [6]. The biochemical structure of the IGF-1 receptor (IGF-1R) is similar to that of the insulin receptor. Free IGF-1 has a low affinity for binding to insulin receptors compared to IGF-IR, stimulating cell proliferation, differentiation, and inhibiting apoptosis. IGF-1R expression is upregulated in HCC, and inhibition of IGF-1 signaling can reduce HCC cell growth and invasion in vitro. A specific inhibitor of IGF-1R signaling significantly inhibits sphere formation of liver cancer cells in vivo [7]. Recent papers indicated that IGF-1 was associated with shorter progression-free survival (PFS) and overall survival (OS) [8] and the elevation of IGF-1/IGF-1R correlated with high tumor recurrence and sorafenib resistance in HCC patients [9–10]. IGF-1 treatment increased epithelial-mesenchymal transition (EMT) markers of migration and metastatic properties in HCC [11–12]. The levels of bioavailable IGF-1 and IGF-II are increased in T2DM patients [13]. Circulating IGF-1 level is a useful predictor of liver reserve in patients with HCC [14]. Overexpressed IGF promotes hepatocarcinogenesis through a phosphoinositide 3-kinase/β-catenin–mediated pathway [15]. In addition, IGF-1 plays an important role in cancer development by regulating angiogenesis, extracellular matrix degradation, liver cancer stem cell self-renewal, tumor invasion, survival, and proliferation [7, 16]. Considering the key role of IGF-1 in the development of liver cancer with pre-existing T2DM, the regulatory mechanism of IGF-1 in liver cells is worth studying.
Autophagy is a conserved catabolic process that maintains cellular homeostasis through the degradation of harmful cytoplasmic components and the recycling of nutrients and plays pivotal physiological and pathological roles in all mammalian cells [17]. Early studies reported the tumor suppressive function of autophagy; moreover, recent studies have revealed a tumorigenesis function [18–19]. The autophagy-related marker microtubule associated protein 1A/1B-light chain 3(LC3) may be a prognostic factor in HCC. Yin Y reported that miR-19-3p can promote autophagy and apoptosis through the AKT/mTOR pathway via targeting IGF-1 [20]. However, it is still unknown whether IGF-1 promotes hepatocarcinogenesis in T2DM through autophagy. The present study aimed to investigate whether increased IGF-1 in T2DM promotes liver cancer growth and metastasis through autophagy. This study may broaden our vision of the mechanisms involving IGF-1 that lead to hepatocarcinogenesis and the role of autophagy in liver cancer and to a worse prognosis.
2Materials and methods2.1Tissue samples from patientsThirty-three HCC patients with T2DM and 33 age-matched HCC patients without T2DM were included in this study. All patients underwent radical liver resection from 2011 to 2016. None of the subjects had other accompanying diseases (e.g., Crohn's disease or Parkinson's disease) that could influence IGF-1 metabolism. All cases had been histologically and clinically diagnosed at Ningbo Medical Center Lihuili Hospital Informed consent was obtained from each patient included in the study and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the Ethics Committee of the Ningbo Medical Center Lihuili Hospital (DXLL2018028). The patient characteristics are listed in Table 1. Glucose levels were under control in all T2DM patients. Tumor tissues were obtained from the non-necrotic regions, and paratumor tissues were obtained from more than 3 cm away from the edge of the tumor.
2.2HepG2 cell culture and treatmentThe human hepatoblastoma cell line HepG2 was purchased from the American Type Culture Collection (HTB-112, ATCC, Rockville, MD, USA). Cells were cultured in Dulbecco's modified Eagle Medium (DMEM) added with 10% fetal bovine serum (FBS, #S11150, ATLANTA Biologicals) in a humidified chamber with 5% CO2 at 37°C.
For in vitro studies, wortmannin (#9951, Cell Signaling) was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) at 2 mM and stored at -20°C and the stock solution was diluted in the medium immediately before being added to the cells. IGF-1 was purchased from Protein Tech and was reconstituted in water at 0.1 mg/mL and stored at -20°C.
2.3Flow cytometryKi-67 staining was used to determine cell proliferation. After trypsin digestion, cells were washed twice with PBS and centrifuged at 1500 rpm for 5 min at 4°C, then resuspended in 1 mL of fixation buffer and incubated at room temperature (RT) for 30 min. Subsequently, cells were centrifuged and resuspended in 100 µL permeabilization wash buffer containing Alexa Fluor® 647 mouse anti-Ki-67 antibody (BD Biosciences, Franklin Lakes, NJ, USA), and then incubated at RT in the dark for 30 min. Finally, 500 µL PBS was added and cells were resuspended for the flow cytometric test.
Cell apoptosis was determined using the Annexin V-PE Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA). Briefly, cells were seeded in six-well plates and then treated with IGF-1 at concentrations of 50, 100 and 200 ng/mL at 37°C for 4 days. Subsequently, cells were washed twice in cold PBS and then resuspended in 1 × binding buffer and 100 μL of the cell suspension was transferred to a 1.5-mL culture tube, to which both 1 μL Annexin V-PE and 1 μL 7-AAD were added. The mixture was incubated for 15 min at RT in the dark. The measurements were then immediately processed with a FACS flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed using FlowJo software.
2.4RNA isolation and real-time reverse transcription-polymerase chain reaction (RT-PCR)Total RNA was extracted from formalin-fixed, paraffin-embedded tumor and paratumor tissues using E.Z.N.A. FFPE RNA kit (OMEGA bio-tek). cDNA was amplified using specific primers with SYBR I Master (Roche). Real-time reverse transcription-polymerase chain reaction (RT-PCR) was conducted using a LightCycler 480 (Roche) and under the following conditions: 95°C for 5 min as an initial denaturation step, then 45 cycles of 94°C for 10 s, 58°C for 20 s, and 72°C for 30 s followed. Beta-actin was used as an external control. The RT-PCR primers were as follows: beta-actin forward: 5’-CCAACCGCGAGAAGATGA-3’; reverse: 5’-CCAGAGGCGTACAGGGATAG-3’; sequestosome 1(SQSTM1)/p62 forward: 5’-TCCAGTCCCTACAGATGCCA-3’; reverse: 5’-AGATGTGGGTACAAGGCAGC-3’: LC3B forward: 5’-AAGGCGCTTACAGCTCAATG-3’; reverse: 5’-CTGGGAGGCATAGACCATGT-3’. -ΔCt (Ct(ACTB)−Ct(target gene)) was used to analyze the relative changes in gene expression.
2.5Western blottingTissue samples (40 mg) were cut into small pieces and 200 µL of protein lysis solution (containing 1% protease inhibitor) was added and homogenized and centrifuged (10,000 rpm, 4°C 10 min), and the supernatant liquid was retrieved. The proteins were extracted from the cells using RIPA buffer (Solarbio Life Science, Beijing, China) for western blotting. Cells were centrifugated at 12,000 × g at 4°C for 20 min in RIPA buffer and then the supernatants were collected. Protein concentrations were determined using the BCA Protein Assay Kit (Cwbio, Beijing, China). Proteins were separated using SDS-PAGE and transferred to a PVDF membrane, which was blocked with 5% fat-free milk for 1 hour. The membranes were incubated with specific primary antibodies overnight at 4°C followed by incubation of the corresponding secondary antibodies for 1 hour at room temperature. Finally, the membrane was incubated with a western blotting detection kit (Advansta, USA). Images were captured by the Tanon 5200 Automated Chemiluminescence Image Analysis System (Tanon, Shanghai, China). Densitometric analysis of the bands was performed using ImageJ software. The proteins were normalized to GAPDH. Primary antibodies used for western blotting are listed in Table 2.
Antibody resources.
Formalin-fixed, paraffin-embedded tissue sections were deparaffinized and then antigen retrieval was performed. Subsequently, the sections were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) in PBS for 5 min, followed by washing with PBS three times. Proteins were blocked for 30 min with 2 mL PBS-0.5% Triton-10% goat serum (Gibco) mixture, then incubated with a rabbit anti-LC3B antibody (#NB600-1384, Novus) at 4°C overnight. After three washes with PBS, sections were incubated with goat anti-rabbit secondary antibody (1:1000, Santa Cruz) in RT for 2 h followed by three washes with PBS. Next, DAPI was used to stain the nuclei, and after rinsing with ddH2O, the sections were observed using a confocal microscope (Olympus Corp, Tokyo, Japan) to quantify the expression of the LC3B protein.
The lentiviral vector with fluorescent-tagged LC3 (GFP-RFP-LC3) was purchased from Gene Chem (Shanghai, China). HepG2 cells were seeded at 1 × 104 cells/mL in an eight-well plate and transfected with GFP-RFP-LC3 lentivirus. Preliminary experiments found that the growth promoting effect was obvious when IGF-1 was stimulated for 24 hours. Cells were treated with IGF-1 for 24 hours after 96 hours of transfection and observed using a confocal microscope. Transfection of the LC3-GFP-RFP lentivirus could reflect cell autophagy by differences in LC3 fluorescence. Both green puncta (GFP) and red puncta (RFP) could be detected when LC3 was expressed in the cytoplasm, which usually represented the initiation of autophagy, but only red puncta were observed in autophagolysosomes, which indicated the terminal period of autophagy.
2.7Cell viability assayThe viability of HepG2 cells was determined by 3-(4-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at the indicated times. Cells were seeded at a density of 2.0 × 103 cells per well in 96-well plates (Corning Costar, Corning, NY, USA) and cultured for 24 hours in 200 µl DMEM supplemented with 10% FBS. Cells were treated with IGF-1 for 1–4 days to determine the temporal correlation and 10 µL of MTT solution (Sigma-Aldrich, St. Louis, MO, USA) solution (5 mg/mL in ddH2O) was added to each well. The plates were then incubated for 3–4 h at 37°C. Intracellular formazan crystals were dissolved by 100 µL DMSO. Cell proliferation was measured at an absorbance of 450 nm with a spectrophotometer (Multiskan MK3; Thermo Fisher Scientific, Waltham, MA, USA). The process was performed referred to the guideline of the MTT kit. The experiments were repeated three times.
2.8Wound healing scratch assayHepG2 cells were seeded in six-well plates by 3–5 × 105 cells per well and grown to 50–80% confluence. The monolayer was scratched with a 200-μL pipette tip. Floating cells were removed by gentle washing with cold PBS, then cultured with DMEM containing 3% FBS and incubated with or without IGF-1 (50, 100, and 200 ng/mL) for 72 hours. The images were captured by an inversion fluorescence microscope and quantified by ImageJ software. All experiments were conducted at least three times.
2.9Transwell assayTranswell invasion assays were performed using Transwell chambers with 8-μm pore size filter membranes (Millipore). The polycarbonate filter was coated with Matrigel (30 μg/well) (BD MatrigelTM Matrix). The chambers were then inserted into 24-well culture plates. Cells were starved overnight in DMEM medium without FBS, and then cells were counted, and single cell suspensions were added to the upper chamber with 5 × 104 cells per well in DMEM without FBS. After being cultured for 24 hours, the upper non-invaded cells of the filter were removed with a cotton swab. The bottom invaded cells were fixed in methanol, stained with 0.1% crystal violet, and observed under a microscope. In migration assays, the filter was not coated with Matrigel, but the other steps were the same as in the invasion assays.
2.10Statistical analysisAll statistical analyzes were performed with GraphPad Prism 6.0 software (GraphPad Software Inc., La Jolla, CA, USA) and SPSS 18.0 software. All values are depicted as mean ± SD and were considered significant if P < 0.05. All data were analyzed using one-way ANOVA with Bonferroni correction, followed by the LSD test for comparison between any two groups.
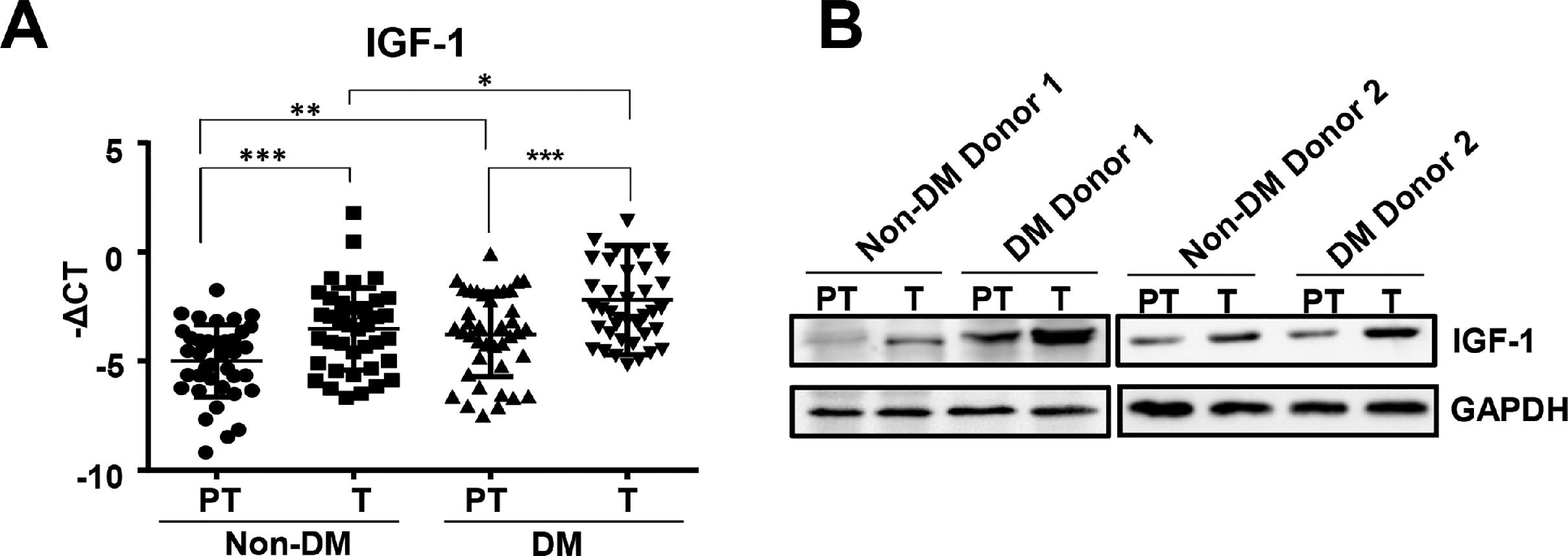
3Results3.1IGF-1 expression increased in liver tumor tissues in HCC patients with diabetesTo study the function of IGF-1 in promoting HCC development in patients with TDM2, we examined the expression of IGF-1 mRNA in HCC tumor and paratumor tissues in patients with or without T2DM. IGF-1 expression was higher in tumor tissues than in paratumor tissues when diabetes was not considered (Fig. 1A). IGF-1 expression was higher in paratumor liver tissues in T2DM patients compared with the subjects without T2DM when HCC was not considered (Fig. 1A). We also observed that IGF-1 expression was also higher in tumor tissues than in paratumor tissues in HCC patients with T2DM (Fig. 1A). Significantly higher expression of the IGF-1 protein was observed in HCC tissues compared to adjacent normal tissues, which was consistent with the mRNA results mentioned above (Fig. 1B).
 The real-time qPCR analysis of transcripts of IGF-1in HCC tumor (T) and para-tumor tissues (PT) in patients with T2DM or not. The IGF-1expression was normalized to ACTB and presented as -ΔCt (n=40). (B) The western blotting analysis of IGF-1in HCC and para-tumor tissues in patients with T2DM or not. The expression was normalized to GAPDH. Error bars represent means ± SD. * P<0.05, ** P<0.01, *** P<0.001.")
IGF-1 is overexpressed in HCC tissues than in para-tumor tissues.
(A) The real-time qPCR analysis of transcripts of IGF-1in HCC tumor (T) and para-tumor tissues (PT) in patients with T2DM or not. The IGF-1expression was normalized to ACTB and presented as -ΔCt (n=40). (B) The western blotting analysis of IGF-1in HCC and para-tumor tissues in patients with T2DM or not. The expression was normalized to GAPDH. Error bars represent means ± SD. * P<0.05, ** P<0.01, *** P<0.001.
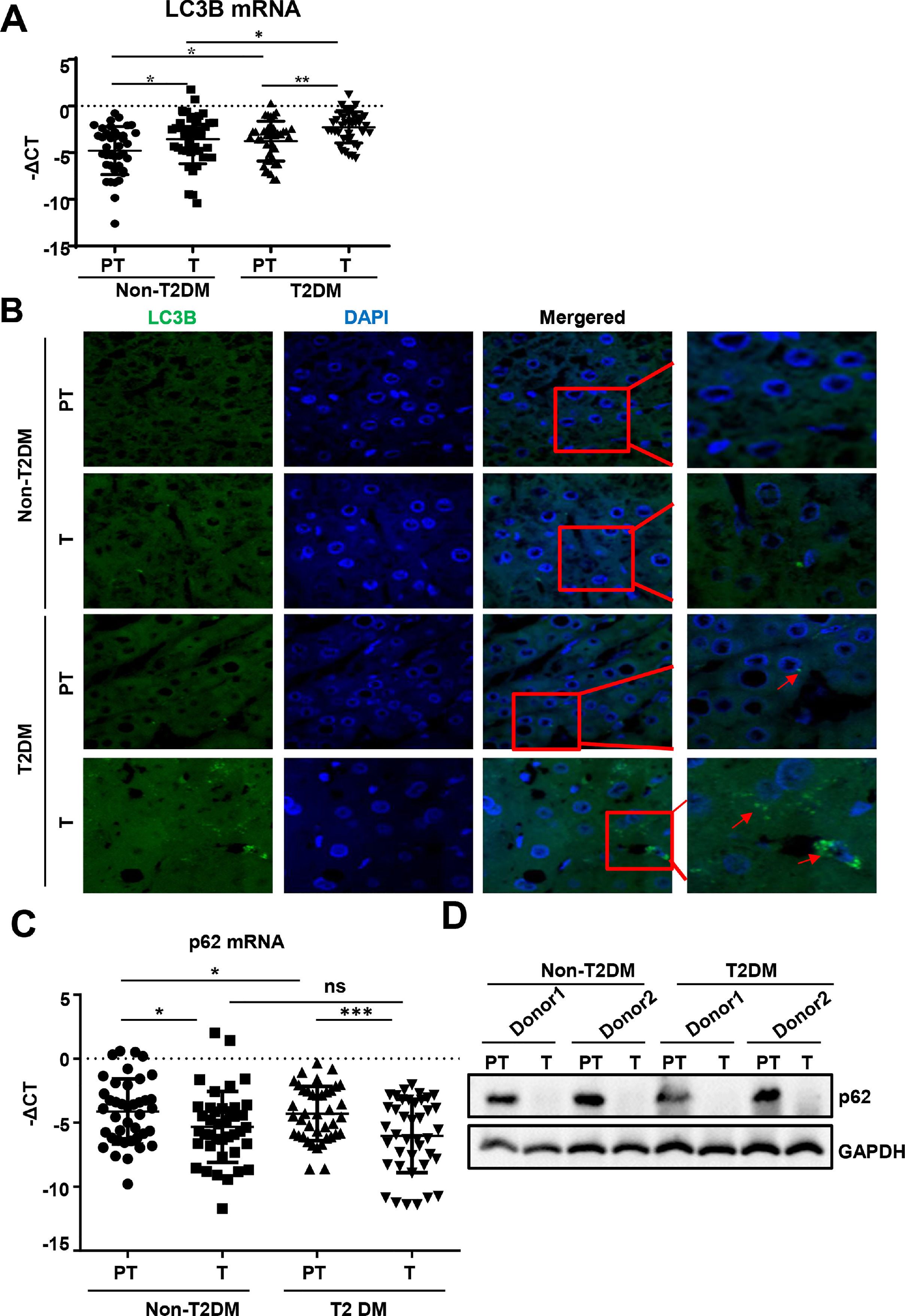
We found that mRNA for the autophagy marker LC3B was higher in liver tumor tissues than in paratumor tissues of patients without T2DM (Fig. 2A). Similar expression trends of LC3B were observed in patients with HCC with T2DM. Additionally, LC3B was higher in liver paratumor tissues of T2DM than in non-T2DM of patients without HCC (Fig. 2A). During autophagy, the LC3B protein is synthesized and undergoes post-translational processing. LC3B is a good marker for monitoring autophagy. We then evaluated LC3B levels in HCC tissues by immunofluorescence and found that LC3B was present in HCC tissues but showed limited expression in normal liver tissues (Fig. 2B). In patients with HCC with T2DM, the levels of LC3B protein in the HCC tissues were even higher than in patients with HCC alone (Fig. 2B).
The real-time qPCR analysis of transcripts of LC3B in HCC and para-tumor tissues in patients with T2DM or not. (B) The immunofluorescence analysis LC3 in HCC and para-tumor tissues in patients with T2DM or not. (C) The real-time qPCR analysis of transcripts of p62 in HCC and para-tumor tissues in patients with type 2 DM or not. (D) The western blotting analysis of p62in HCC and para-tumor tissues in patients with DM or not. The expression was normalized to GAPDH. Error bars represent means ± SD. The p62 transcripts expression was normalized to ACTB and presented as -ΔCt. * P<0.05, * P<0.51, *** P<0,*** P<0.001.001.")
Autophagy is upregulated in HCC tissues than in para-tumor tissues.
(A)The real-time qPCR analysis of transcripts of LC3B in HCC and para-tumor tissues in patients with T2DM or not. (B) The immunofluorescence analysis LC3 in HCC and para-tumor tissues in patients with T2DM or not. (C) The real-time qPCR analysis of transcripts of p62 in HCC and para-tumor tissues in patients with type 2 DM or not. (D) The western blotting analysis of p62in HCC and para-tumor tissues in patients with DM or not. The expression was normalized to GAPDH. Error bars represent means ± SD. The p62 transcripts expression was normalized to ACTB and presented as -ΔCt. * P<0.05, * P<0.51, *** P<0,*** P<0.001.001.
During autophagy, the polyubiquitin-binding protein p62, also called SQSTM1, is incorporated into the completed autophagosome and is then degraded by autophagy [21]. We found that mRNA and protein for p62 expression was lower in liver tumor tissues compared to paratumor tissues and that this trend existed regardless of the accompanying T2DM (Fig. 2C and D).
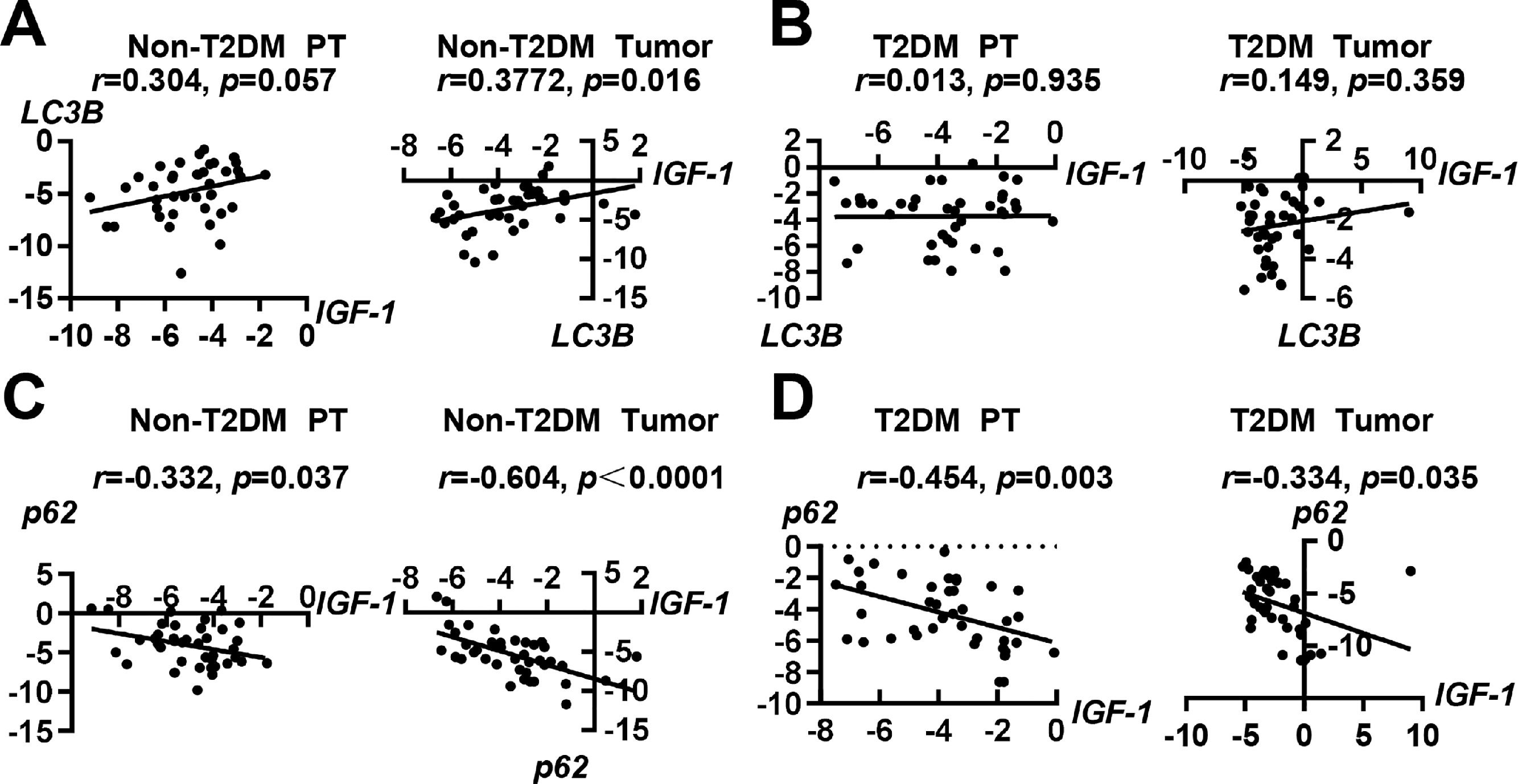
3.3Correlations of IGF-1 with autophagy-related molecules LC3B and p62To investigate whether IGF-1 was correlated with autophagy-related molecules LC3B and p62, we performed a correlation analysis. In patients with HCC without T2DM, the expression of IGF-1 mRNA was positively correlated with LC3B in tumor tissues, but the correlation was not significant in paratumor tissues (Fig. 3A). In patients with HCC with T2DM, the expression of IGF-1 and LC3B showed no correlation in either tumor tissues or paratumor tissues (Fig. 3B). Interestingly, the expression of IGF-1 mRNA was negatively correlated with p62 in both tumor tissues and paratumor tissues regardless of accompanying T2DM (Fig. 3C and 3D).
 Correlation analysis of IGF-1 with LC3B in para-tumor and HCC tissues in patients without T2DM. (B) Correlation analysis of IGF-1 with LC3B in para-tumor and HCC tissues in patients with T2DM. (C) Correlation analysis of IGF-1 with p62 in para-tumor and HCC tissues in patients without T2DM. (D) Correlation analysis of IGF-1 with p62 in para-tumor and HCC tissues in patients with T2DM.")
Correlation analysis of IGF-1 with LC3B and p62 in HCC and para-tumor tissues in patients with T2DM or not. (A) Correlation analysis of IGF-1 with LC3B in para-tumor and HCC tissues in patients without T2DM. (B) Correlation analysis of IGF-1 with LC3B in para-tumor and HCC tissues in patients with T2DM. (C) Correlation analysis of IGF-1 with p62 in para-tumor and HCC tissues in patients without T2DM. (D) Correlation analysis of IGF-1 with p62 in para-tumor and HCC tissues in patients with T2DM.
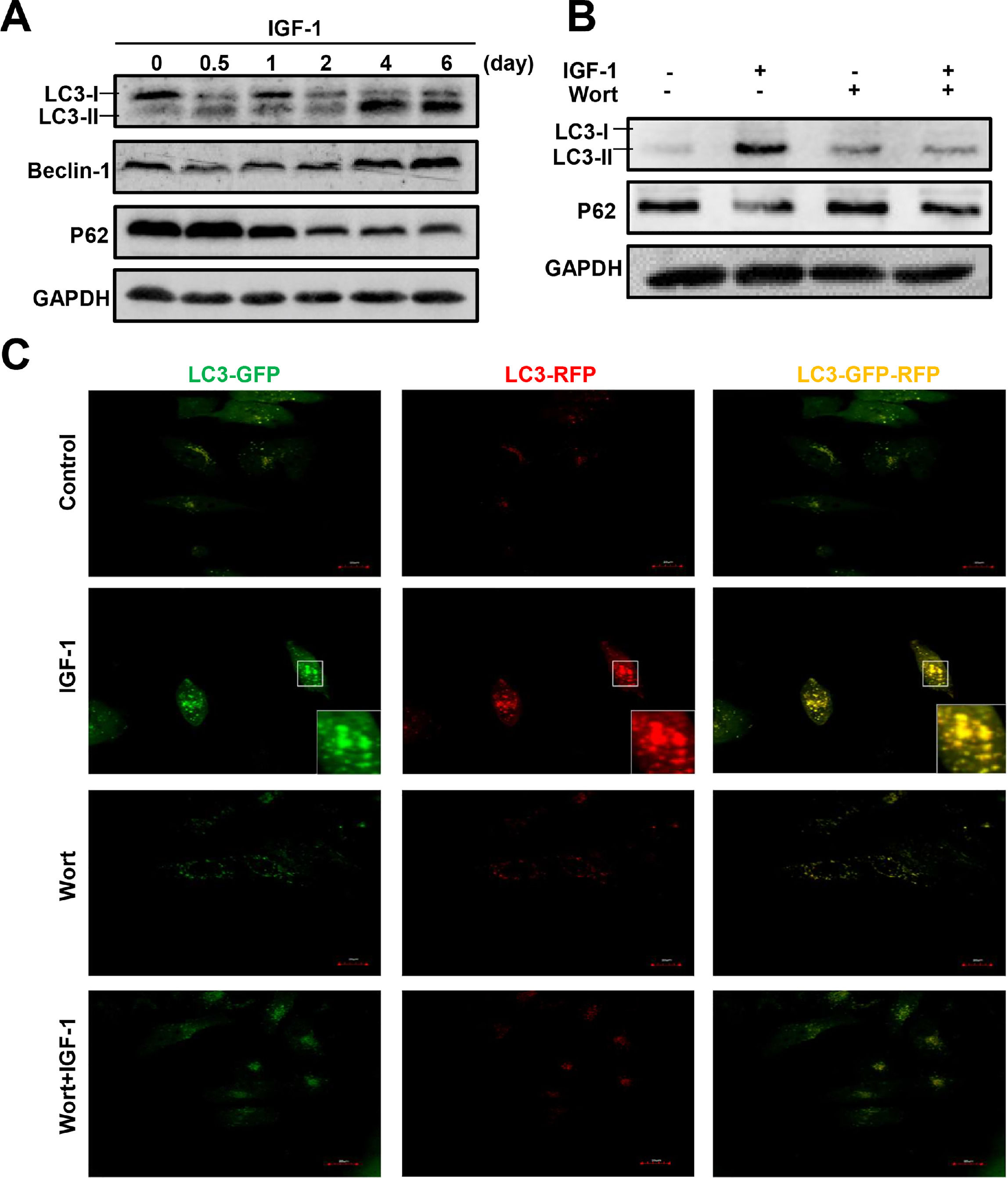
To determine whether IGF-1 affects hepatic cell autophagy, we cultured a human hepatoblastoma cell line HepG2 cells in the presence of IGF-1 for different periods of time, from 12 hours to 6 days. Autophagy-related proteins were then measured by western blotting. The LC3B-I transforms to LC3B-II indicating the formation of autophagosomes. IGF-1 promoted autophagy when administered for 4 or 6 days (Fig. 4A). The expression of the autophagy marker Beclin-1 increased and p62 was down-regulated (Fig. 4A). As shown in Fig. 4B, wortmannin, a known autophagy inhibitor, inhibited IGF-1-induced autophagy. Under normal circumstances, autophagy was not activated in HepG2 cells; as shown by the lack of staining in control cells. The green and red puncta markedly increased when cultured with IGF-1, which reflected the autophagy stimulation by IGF-1. Wortmannin pretreatment inhibited autophagy at initiation; therefore, green and red puncta were absent in the 'Wort + IGF-1′ and 'Wort' groups, which meant that both the autophagosome and the autophagolysosome were inhibited. The results, as shown in Fig. 4C, were consistent with those from the western blotting (Fig. 4B).
 The western blotting analysis of autophagy related proteins LC3-II, Beclin-1, and p62 in HepG2 cells treated with IGF-1 (200 ng/ml) for 0.5-6 days. (B) Western blotting of LC3-II and p62 in HepG2 cells treated with IGF-1 (200 ng/ml) and Wortmannin (2μM/ml) for 24 h. (C) Fluorescence of GFP-RFP-LC3 lentivirus transfected HepG2 cells, which were treated with IGF-1(200 ng/ml) for 24 h. GFP (green fluorescence) represented autophagosomes that had not fused with lysosomes. RFP (red fluorescence) represented autophagolysosome. When the green and red fluorescence existed at the same time, it was yellow which meant autophagy induced.")
IGF-1 promotes HepG2 cell autophagy and the activation could be inhibited by Wortmannin.
(A) The western blotting analysis of autophagy related proteins LC3-II, Beclin-1, and p62 in HepG2 cells treated with IGF-1 (200 ng/ml) for 0.5-6 days. (B) Western blotting of LC3-II and p62 in HepG2 cells treated with IGF-1 (200 ng/ml) and Wortmannin (2μM/ml) for 24 h. (C) Fluorescence of GFP-RFP-LC3 lentivirus transfected HepG2 cells, which were treated with IGF-1(200 ng/ml) for 24 h. GFP (green fluorescence) represented autophagosomes that had not fused with lysosomes. RFP (red fluorescence) represented autophagolysosome. When the green and red fluorescence existed at the same time, it was yellow which meant autophagy induced.
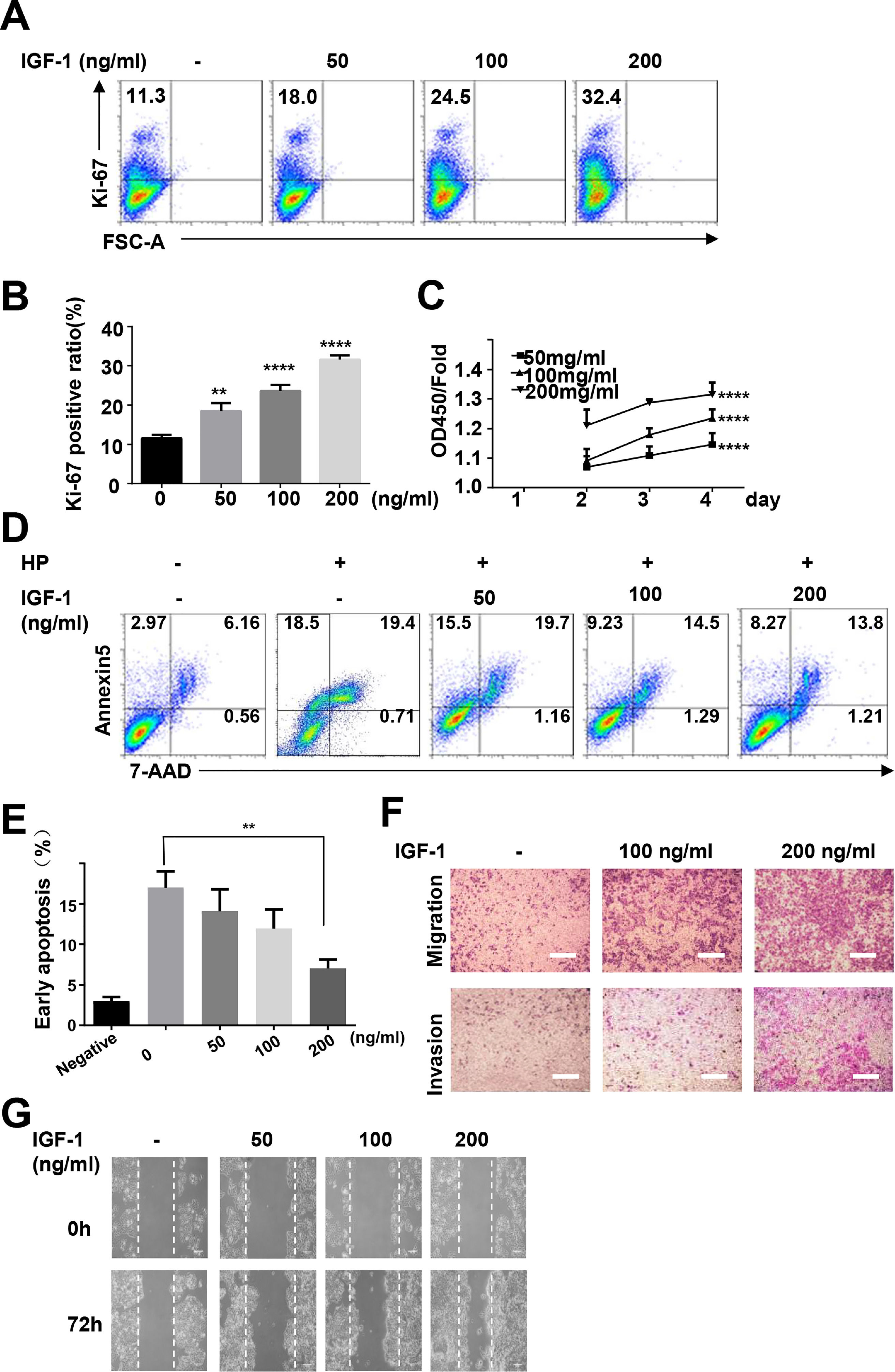
To study the possible functions of IGF-1 in liver cancer, we treated HepG2 cells with IGF-1 to examine the effects of IGF-1 on cell growth. IGF-1 promoted HepG2 cell growth in a dose-dependent manner (Fig. 5A–C). The proliferation-promoting effect of IGF-1 was significant when cells were cultured for 4 days. Next, we investigated the effects of IGF-1 on apoptosis. Considering that apoptosis was detected in only 2.97% of the cells at baseline, we cultured cells with hydrogen peroxide to stimulate apoptosis after pretreating cells with IGF-1 for 4 days. After treatment with hydrogen peroxide for 5 minutes, apoptosis was detected. Hydrogen peroxide-induced apoptosis was decreased when cells were pretreated with IGF-1 (Fig. 5D and E). Next, we examined whether IGF-1 was able to increase the migration and invasion abilities of HepG2 cells. Cells were first treated with different concentrations of IGF-1 and then subjected to Transwell assays to assess migration and invasion. As shown in Fig. 5F, IGF-1 enhanced cell migration and invasion in HepG2 cells in a concentration-dependent manner when administered for 4 days. Wound healing scratch assays were also performed to examine the effects of IGF-1 on cell migration. IGF-1 treatment accelerated cell migration in HepG2 cells, as shown in Fig. 5G.
 Flow cytometry of Ki-67 expression in HepG2 cells. Data are representative of three experiments. (B) Statistical analysis of the percentage of Ki-67+ HepG2 cells. (C) MTT assays of IGF-1 (200 ng/ml) treated HepG2 cell proliferation in 1-4 days. (D) Flow cytometry of Annexin V/7-AAD in HepG2 cells treated with IGF-1 for 4 days then treated with hydrogen peroxide (HP) for 5 min. (E) Statistical analysis of the percentage of Annexin V+ 7-AAD- HepG2 cells. (F) Transwell assay of IGF-1 treated HepG2 cells. The cells on the lower surface of the chambers were stained and imaged. Scale bar, 20 μm. (G) Healing scratch assay of IGF-1 treated HepG2 cells. HepG2 cells were incubated with various concentrations of IGF-1 in 6-well plates and the migration of cells was assessed by wound healing scratch assay. The migratory distance was calculated at 72 h in HepG2 cultures before complete wound closure. Data are representative of three experiments. Data represent means ± SD.** P<0.01, **** P<0.0001.")
IGF-1 promotes HepG2 cell proliferation, migration and protected HepG2 cells from apoptosis.
HepG2 cells were treated with IGF-1 at the concentrations of 50, 100 and 200 ng/ml for 4 days. (A) Flow cytometry of Ki-67 expression in HepG2 cells. Data are representative of three experiments. (B) Statistical analysis of the percentage of Ki-67+ HepG2 cells. (C) MTT assays of IGF-1 (200 ng/ml) treated HepG2 cell proliferation in 1-4 days. (D) Flow cytometry of Annexin V/7-AAD in HepG2 cells treated with IGF-1 for 4 days then treated with hydrogen peroxide (HP) for 5 min. (E) Statistical analysis of the percentage of Annexin V+ 7-AAD- HepG2 cells. (F) Transwell assay of IGF-1 treated HepG2 cells. The cells on the lower surface of the chambers were stained and imaged. Scale bar, 20 μm. (G) Healing scratch assay of IGF-1 treated HepG2 cells. HepG2 cells were incubated with various concentrations of IGF-1 in 6-well plates and the migration of cells was assessed by wound healing scratch assay. The migratory distance was calculated at 72 h in HepG2 cultures before complete wound closure. Data are representative of three experiments. Data represent means ± SD.** P<0.01, **** P<0.0001.
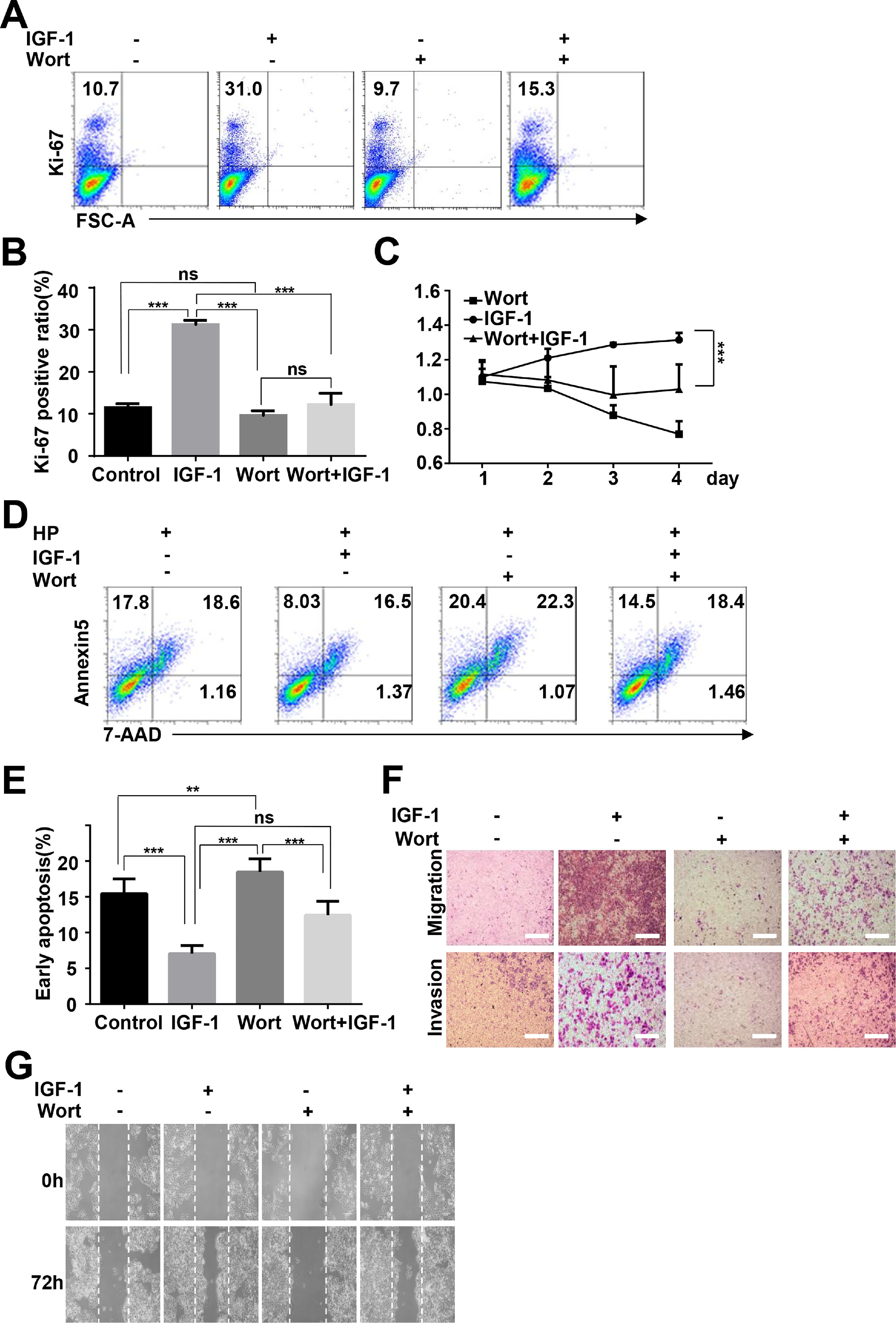
To determine whether IGF-1 promotes HepG2 cell proliferation and migration through regulation of autophagy, we inhibited autophagy by preculturing HepG2 cells with wortmannin and then added IGF-1 for 4 days. Cell proliferation, invasion, migration, and apoptosis were measured as indicated above. After the cells were pretreated with wortmannin, the proliferation-promoting effect of IGF-1 was not observed in HepG2 cells (Fig. 6A–C). Moreover, wortmannin reversed apoptosis inhibition (Fig. 6D and E). Furthermore, IGF-1 improved cell migration, while invasion in HepG2 cells was reversed by wortmannin (Fig. 6F and G).
 for 1 h and then treated with IGF-1 (200 ng/ml) for 4 days. Ki-67 staining (A and B) and MTT assays (C) for HepG2 cell proliferation. (D) Cell apoptosis was determined by Annexin V-PE/7-AAD double staining. (E) Statistical analysis of Annexin V+ 7-AAD- HepG2 cells. Transwell assays (F) and wound healing assays (G) showed the difference of migration and invasion of HepG2 cells between pre-cultured with wortmannin and only cultured with IGF-1. Data are representative of three experiments. Data represent means ± SD.** P<0.01, *** P<0.001.")
IGF-1 promotes HepG2 cell proliferation and migration through increasing autophagy.
HepG2 cells were pre-treated with Wortmannin (2 μM/ml) for 1 h and then treated with IGF-1 (200 ng/ml) for 4 days. Ki-67 staining (A and B) and MTT assays (C) for HepG2 cell proliferation. (D) Cell apoptosis was determined by Annexin V-PE/7-AAD double staining. (E) Statistical analysis of Annexin V+ 7-AAD- HepG2 cells. Transwell assays (F) and wound healing assays (G) showed the difference of migration and invasion of HepG2 cells between pre-cultured with wortmannin and only cultured with IGF-1. Data are representative of three experiments. Data represent means ± SD.** P<0.01, *** P<0.001.
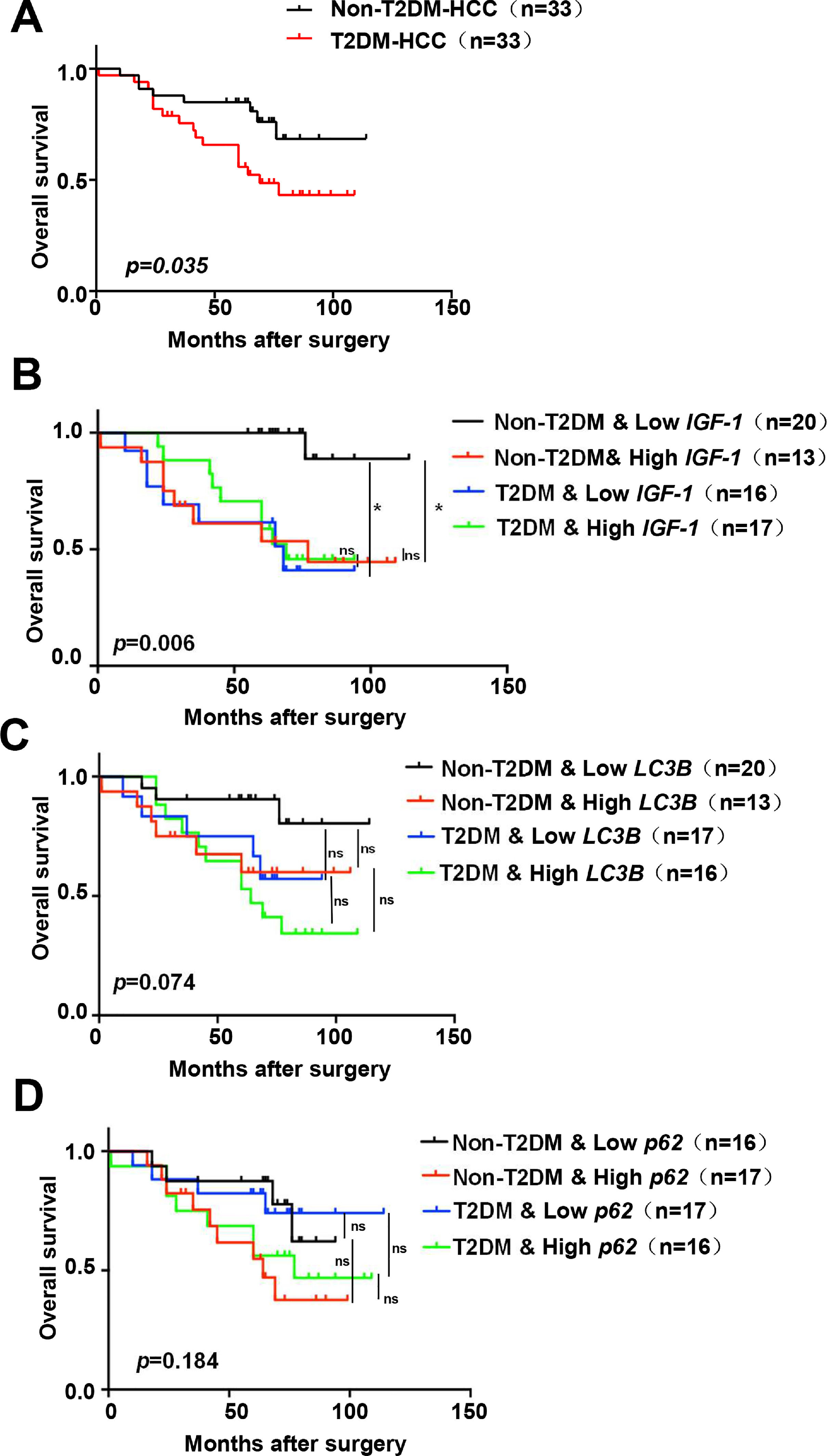
Based on the above results, all patients were divided into groups according to the expression of IGF-1, LC3B, p62, and whether they had diabetes. Survival analysis indicated that the T2DM group had a worse prognosis than the non-T2DM group (p=0.035) (Fig. 7A). The non-T2DM/Low IGF-1 expression group had the best prognosis, which was significantly different compared to the Non-T2DM/High IGF-1 expression group (p=0.001, 0.002) (Fig. 7B). The patients were grouped according to the expression of LC3B and T2DM status; the non-T2DM&Low LC3B expression group had the best prognosis, while the T2DM/High LC3B expression had the worst prognosis. In both the T2DM and the non-DM groups, the prognosis of the high LC3B expression groups was poorer compared to the low LC3B expression groups, but the difference was not statistically significant (Fig. 7C). There was no obvious correlation between p62 and prognosis (Fig. 7D).
 Kaplan–Meier analyses of overall survival in HCC patients with or without T2DM (n=33 for each group). (B-D) Kaplan–Meier analyses of overall survival in all of the HCC patients (with or without T2DM) in the study cohort classified by expression of mRNA for IGF-1, LC3B and p62. (B) Overall survival in T2DM and non-T2DM HCC patients classified into IGF-1 low and high groups according to the median. (C) Overall survival in T2DM and non-T2DM HCC patients classified into LC3B low and high groups according to the median. (D) Overall survival in T2DM and non-T2DM HCC patients classified into p62 low and high groups according to the median. * P<0.05.")
Kaplan–Meier survival analysis of IGF-1, LC3B and p62 expression in HCC patients
(A) Kaplan–Meier analyses of overall survival in HCC patients with or without T2DM (n=33 for each group). (B-D) Kaplan–Meier analyses of overall survival in all of the HCC patients (with or without T2DM) in the study cohort classified by expression of mRNA for IGF-1, LC3B and p62. (B) Overall survival in T2DM and non-T2DM HCC patients classified into IGF-1 low and high groups according to the median. (C) Overall survival in T2DM and non-T2DM HCC patients classified into LC3B low and high groups according to the median. (D) Overall survival in T2DM and non-T2DM HCC patients classified into p62 low and high groups according to the median. * P<0.05.
In this study, we found that IGF-1 was up-regulated in HCC tumor tissues compared to normal tissues, and that IGF-1 promoted HepG2 cell proliferation and metastasis, but inhibited apoptosis. Ma et al. reported that IGF-1 induced the proliferation and migration of human HCC cells [15]. However, Yao et al. reported that a low preoperative circulating IGF-1 level was significantly associated with an increased risk of early recurrence of HCC [14]. Lei et al. revealed that IGF-1 promoted the growth and metastasis of HCC cell lines. Furthermore, HCC cells grew much faster in diabetic mice [22]. In the present study, the duration of diabetes in most patients with HCC and T2DM was longer than 3 years, and their glycemic indices were under control. Therefore, even when the glucose levels of patients with T2DM were normal, differences in cytokine expression still influenced tumor cell survival.
Our results revealed that IGF-1 was upregulated in both diseases and promoted tumor cell survival and metastasis. These data suggest that the chronic effect of T2DM on HCC could be due to inflammatory factors such as IGF-1 but not to blood glucose level. Accelerated colon tumor growth was found in a mouse model of T2DM, db/db mice, which has high levels of endogenous insulin and IGF-1 [20]. Many laboratory studies have found that IGF-1 promotes glucose uptake in peripheral tissues and suppresses hepatic glucose production [23–25]. Whether the role of IGF-1 in maintaining glucose homeostasis correlates with its role in tumor development remains to be discovered.
The dysregulation of autophagy is associated with viral hepatitis, non-alcoholic fatty liver disease, alcoholic liver disease, fibrosis, cirrhosis, and liver cancer [26]. Autophagy is activated in tumor cells under various types of stress and promotes cancer cell survival because autophagy plays an important role in both the quality control of organelles and the supply of amino acids and fatty acids to cells [27]. Cancer cells increase their autophagic activity to survive hostile microenvironments [28]. Autophagy promotes HCC invasion through the activation of epithelial–mesenchymal transition [29] and SQSTM1 overproduction in mouse liver cancer precursor cells was found to drive HCC tumorigenesis via activation of both mTORC1 and NRF2 [30]. However, the functions of autophagy in HCC are more complex as a tumor suppressive role of autophagy has also been reported. Beclin-1 heterozygous disruption in mice leads to increased frequency of malignancies and promotes the development of premalignant lesions [31]. In mice with liver-specific knockout of Atg7 and p62, liver adenoma growth was strongly suppressed [32]. T2DM and HCC are chronic inflammatory diseases in which inflammatory factors are dysregulated [2]. To date, few reports have evaluated the effects of IGF-1 on autophagy in diseases. In the present study, although we were unable to directly evaluate autophagic flux in human HCC samples, we measured the expression of Beclin-1 and LC3, which have been widely used to quantify autophagosomes. These results strongly suggested that autophagy was altered in HCC compared to normal cells.
Our study also revealed that p62 expression was lower in HCC cells than in normal cells, suggesting an accelerated autophagic flux. In our in vitro study, we found that autophagic flux and autophagy-related protein expression were altered in HCC cells when exposed to IGF-1. The results of the in vivo experiments suggested that IGF-1 enhanced autophagy in HCC combined with T2DM. We also determined that IGF-1 promoted HCC cell growth and migration and decreased apoptosis, and that IGF-1 activated HCC cell proliferation and migration could be blocked by inhibiting autophagy. This may imply that, similar to other tumors [33], autophagic degradation provides the energy and materials for HCC tumor cells growth. In addition to autophagy, pleiotropic growth factor IGF-1 may affect hepatocarcinogenesis through other pathways or molecules such as BDNF [34–37]. Furthermore, autophagy may participate in downstream reactions induced by chronic inflammation or inflammatory factors. Therefore, autophagy might become a potential therapeutic target. To assess the predictive role of IGF-1 in prognosis, we followed patients over 5–8 years. Survival analysis showed that the prognosis of HCC complicated by diabetes is significantly worse, and our results confirmed that diabetes was a risk factors for the poor prognosis of HCC. Increased expression of IGF-1 was often observed in diabetic patients and was associated with a poor prognosis of HCC. Recently, researcher identified the autophagy-associated genes(EIF2S1, BIRC5, SQSTM1,ATG7, HDAC1, and FKBP1A) correlated with HCC prognosis and constructed a corresponding prognostic risk model [38]. In our paper, the prognosis of patients with low LC3B was better than the ones with high LC3B levels. The non-significant difference of autophagy-related LC3B and p62 in HCC prognosis in our study may be due to the deficient number of the patients. A larger number of patients should be included in further studies.
ConclusionsIn conclusion, the present study demonstrated that IGF-1 activates autophagy, which in turn promotes HepG2 cell survival and metastasis, thus affecting the prognosis of liver cancer. IGF-1 may be the key inflammatory factor that leads to the high risk of HCC, especially that accompanied by T2DM, therefore IGF-1 could be a potential therapeutic target for HCC patients,especially with T2DM.
Availability of data and materialsThe datasets supporting the conclusions of this article are included within the article.
Authors contributionsFW and CL conceived and designed the study. YS conducted experiments and drafted the article. JW, ML and SY collected and analyzed the data. SW, JH and SB revised the manuscript for important intellectual content. SB reviewed and approved the final version of manuscript. All authors have read and agreed to the published version of the manuscript.
Declaration of interestNone.
This work was supported by the National Natural Science Foundation of China (81370165 to SB; 81501421 to FW), the Zhejiang Provincial Natural Science Foundation of China (LQ16H100001 to FW), Zhejiang Medicine and Health Science and Technology Project (2019ZD047 to SW), the Natural Science Foundation of Ningbo Municipality (2021J100 to FW).



