



HepG2 human hepatoma cells were incubated for 24 or 48 h with various concentrations of YHK solution. After 24 h incubation, cell proliferation and cytotoxicity were determined by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2Htetrazolium (MTT) assay. Cytotoxicity or necrosis was expressed as lactate dehydrogenase (LDH) release. After exponential growth phase HepG2 cells were treated with different doses of YHK and apoptosis was assessed by using an Annexin V-FITC kit. Further, oxidative stress was measured by dichlorofluorescein-diacetate (DCFH-DA) assay. As compared to control, YHK-treated cultures showed a significant time-course decrease of the proliferation rate of HepG2 cell growth (p < 0.01). This is likely to be due to an enhanced cytotoxicity (MTT and LDH tests) (p < 0.001). On the other hand, YHK showed in vitro to significantly enhance the oxidative stress of HepG2 cell (p < 0.01) while also markedly increasing apoptosis at 72 h with cells G2/M phase arrest (p < 0.01). These data suggest that YHK seem to modulate the extrinsic and intrinsic regulators of apoptosis and sensitize tumour cells to apoptosis. These preliminary data are worth interest when considering that this nutraceutical has been shown in vitro and in vivo to exert protective anti-tumour effect by redox status-modulating and immuno-regulatory actions. Given its lack of toxicity so far reported, such natural product might represent an effective nutritional supplement in a number of pathological conditions where a chemo-preventive strategy is planned.
Human hepatocellular carcinoma (HCC) is one of the major causes of death worldwide,1-4 and, besides some limited cases which are amenable to transplantation,5,6 surgical resection is the only curative therapy. However, this poses limitations for patients with multiple type or metastatic tumours. Thus, the search for effective chemotherapeutic agents remains and important goal aimed to improve the survival rate of patients with advanced or recurrent HCC after surgical treatment. On the other hand, HCC is usually insensitive to the available chemotherapeutics given its expression of mult-idrug resistance gene. This prompts the need for the evaluation of new active drugs against HCC. It has been shown that both cell proliferation and apoptosis are involved in HCC7,8 and, in particular, cell proliferation seem to increase as HCC progresses into more poorly differentiated forms.9,10 In recent years we have been doing a number of experimental and clinical tests suggesting the potent hepatoprotective properties of a natural compound, i.e. YHK (panax pseudo-ginseng, Eucommia Ulmoides, polygonati rhizome, glycyrrhiza licorice, panax ginseng, Kyotsu Jigyo, Tokyo, Japan) besides its safety profile.11,12 Moreover, quite recently this compound has been shown in vivo experimental hepatocarcinogenesis model to significantly inhibit the malignant transformation.13 This study was thus aimed to investigate its potential as a growth inhibitor and apoptosis enhancer in human HepG2 hepatocellular cell line in order to provide the theoretical basis for clinical application in patients with HCC.
Material and methodsCells and chemicalsHepG2 human hepatoma cells were purchased from American Type Culture Collection (ATCC). Eagles minimum essential medium (MEM, a-modified), Dichlorofluorescein-diacetate (DCFH-DA), dimethyl sulfoxide (DM80) and AAPH were purchased from Sigma. A stock solution of DCFH-DA (20jM) was prepared in DMSO and stored at -20°C for up to 1 month. AAPH (166,4mM) working stock solution was made fresh daily by dissolving in an appropriate amount in PBS. AAPH (the free radical generator in this system) was kept on ice until used.
Cell cultureCells were cultured in Dulbecco’s MEM (1x) medium supplemented 2mM glutamine, 10% fetal calf serum, 40U/mL streptomycin and 50U/mL penicillin at 37°C in a humidified atmosphere of 950 mL/L air and 50 mL/L CO2, as described elsewhere.14 Cells were maintained as monolayer cultures and media was changed every two days. Before starting the test treatment, the cells were grown to 80-90% confluency and synchronized by incubating in the basal medium (100% MEM) for 24 h so that confluent HepG2 cell cultures were used for all experiments.
Preparation of culture media containing test compoundHot-water-extracted YHK was prepared and concentrated to one gram of compound in one millilitre of distilled water. After dissolved and mixed at 37°C for 30 min in DMEM, the concentration of YHK was adjusted to 100 mg/mL. This solution was centrifuged (1,200 X g, 30 min) to remove insoluble ingredients. The supernatant was sequentially passed through 0.45 μm and 0.22 μm filters sterilization.
Cell viability, proliferation and citotoxicity assayAfter having checked cell viability by colorimetric assay set at 490 nm, samples containing 200 μL cell suspension were dispersed with 0.02% edetic acid to prepare the suspension of a cell density of 3 × 107/L. Then cell suspension was fractionated into a 96-well flat-bottomed microtiter plates in a total volume of 100 mL per well and incubated in a 5% CO2 atmosphere at 37° for 4 h. Cultures were incubated for 24 with various concentrations (0-200 mg/L) of YHK solution. After 24 h incubation, cells were added with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2Htetrazolium (MTT) solution (15), 5 g/L (20 mL per well) and 25 μmol/L phenazine methosulfate. After another 4-h incubation, the supernatant were discarded and isopropanol hychloride 0.04 mol/L were added (100 mL per well). Then the suspension was vibrated on a micro-vibrator for 5 min and the absorbance (A) of soluble formazan produced by cellular reduction of MTS was measured at 570 nm using an ELISA reader. The cell inhibitory ratio was calculated with the following formula: Inhibitory ratio = (1-A Treated group /AControl group) × 100%. All assays were completed in triplicate results were confirmed by direct cell counting using a hemocytometer. Moreover, cells were labeled with 3.7 × 104 Bq of 3H-thymidine, and incorporation into DNA was quantitated in a scintillation counter 24 h later.
Cytotoxicity were determined by lactate dehydrogenase (LDH) release from cells according to the decrease in the absorbance at 340 nm for NADH disappearance. Briefly, after incubation with various concentrations of YHK for 24 h, the cell culture supernatant and medium (100 μL) were mixed with 900 μL of modified Krebs-Henseleit buffer (118 mmol/L NaCl, 24 mmol/L NaHCO3, 1 mmol/L KH2PO4, 4.8 mmol/L KCl, 3 mmol/L CaCl2, 0.8 mmol/L MgPO4, pH 7.4), 20 mg/L bovine serum albumin, 1.36 mmol/L sodium pyruvate and 0.2 mmol/L NADH. The percentage of LDH release was obtained by calculationg LDH activity in medium divided by that in both cell culture supernatant and medium × 100%.16 Cells without addition of YHK extract were incubated with 10 mL/L ethanol and 10 mL/L sorbitol solution as the control group. The cells and medium were collected. Protein contents in cells and medium were measured by the modified method of Lowry et al.17
Assessment of cell cycle progression and of apoptosis.Cells were seeded in a flask at a density of 1 ×106 cells/flask. After 24 h, at a final concentration of 200 mg/L YHK was added to the respective flask and incubated for 24, 48 or 72 h. Cells were trypsinized, harvested, and fixed in 1 mL 80% cold ethanol in test tubes and incubated at 4°C for 15 min. After incubation, cells were centrifuged at 1,500 rpm for 5 min and the cell pellets were re-suspended in 500 μl propidium iodine (10 μg/mL) containing 300 μg/mL RNase (Sigma, MO, USA). Then cells were incubated on ice for 30 min and filtered with 53 μm nylon mesh. Cell cycle distribution was calculated from 10,000 cells using FACS caliber (Becton Dickinson, CA, USA). Apoptosis was assessed by using an Annexin V-FITC kit binding assay and analyzed by cytometry using a fluorescence-activated cell sorter (FACS). Cell suspensions (2 mL), prepared as above mentioned, were re-suspended in binding buffer (10 mM Hepes/NaOH, pH7.4, 140 mM NaCl, 2.5 mM CaCl2) and Annexin V-FITC was added and incubated for 30 min in 96-well plates at room temperature in the dark. After 24 h of incubation at 37°C with 5% CO2, YHK solutions were added. After a further 72 h incubation, cells were collected by centrifugation and stained in Tris-buffered saline containing 50 μg/mL PI, 10 μg/ mL RNAse of DNAse and 0.1% Igepal CA-630 for 1h at 4°C. FACS analysis was carried out by a flow cytometer at 488 nm. Data from 4.000 cells were collected and analyzed in triplicate.
Assessment of oxidative stress in HepG2 cells by DCFH-DA assayAfter checking the viability of the cells by trypan blue exclusion, these were plated (10.000 cells/well) into 96-well plates 12 hours before starting the test. On the experimental day, hydrophilic YHK extracts were thawed and diluted with PBS (pH 7.3). After 12 hours of incubation, the medium was removed and cells were washed with PBS and then treated with 50 μL PBS, 0-200 mg/L YHK extract or 50 jul PBS for control. 50 μL DCFH-DA (final concentration: 5 μLM) and 50 ßL AAPH (final concentration: 41.6 mM) for a total volume of 200 μL/well were employed. Fluorescence was measured at once and every 5 min after adding AAPH for 30 min using a fluorescence plate reader (excitation filter 495/20 nm, emission filter 528/20 nm). All samples were performed in triplicate by randomly sampling within each plate. Data were expressed as fluorescence intensity/well. After the 30 min assay, cells were washed with PBS and their protein analyzed as follows. Briefly, cells were lysed with 50 μL of a 0.8% filtered digitonin solution to each well followed by incubation for 10 min at 37°C. Then, 200 μL Bio-Rad dye were added to each well and intensity of the dye was read at 595 nm on a microplate reader. Results were expressed as mg protein/mL for each well and bovine serum albumin was used as a reference standard.
Final data were expressed either as fluorescence intensity/mg protein or as a mean percent decrease in fluorescence intensity (ROS production/mg protein ± standard deviation compared to the control (DCFH-DA and AAPH without YHK extract) at the 30 min time period. Data were calculated by using the following formula: (100c - (Fb30/ Fc30 *100), where 100c represents 100% fluorescence intensity/ mg protein/30 min displayed by control, Fb30 is the fluorescence intensity/mg protein in the presence of YHK extract at 30 min and Fc30 stands for the fluorescence intensity/mg protein of the control at 30 min. The data were standardized and their variation controlled by dividing the fluorescence intensity/mg protein of all samples by the fluorescence intensity/mg protein of the control sample each time.
Statistical analysisData are expressed as x±s. Data were analyzed by oneway ANOVA and Fisher’s least significant difference test was used to make post-hoc comparisons if the treatment effect was demonstrated. Differences were considered significant when P < 0.05. Means of three samples per plate were used for calculations.
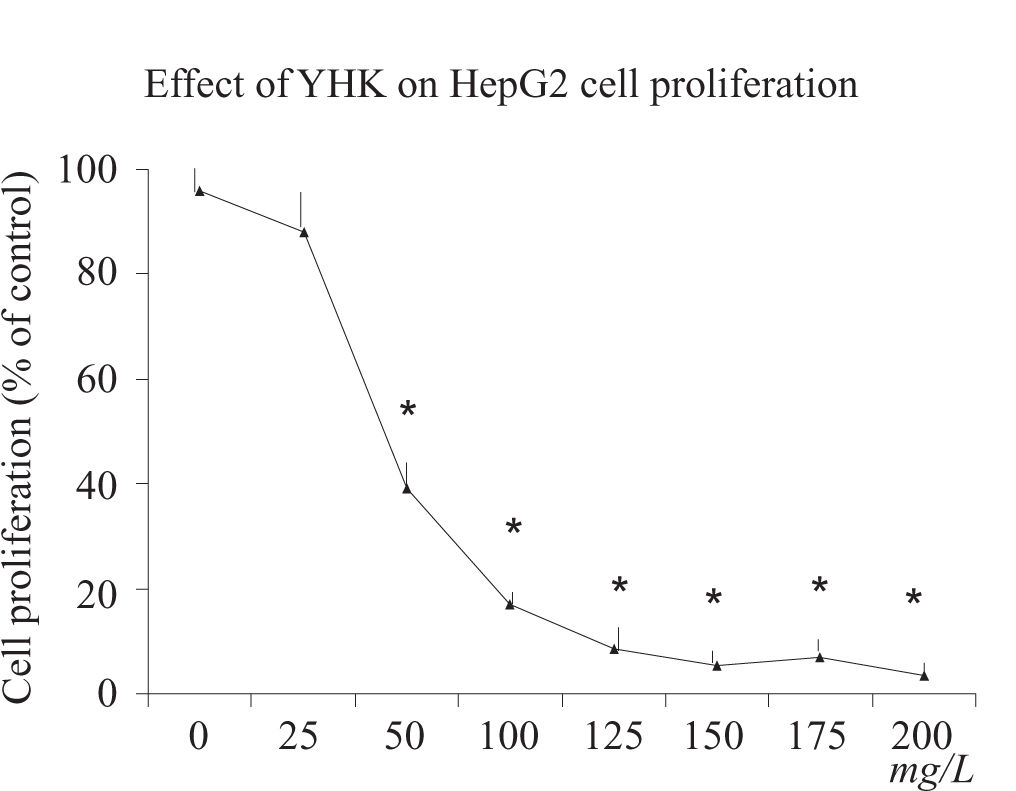
ResultsCell proliferation assayAfter 24 h incubation, YHK (25-200 mg/L) significantly inhibited cell growth (p > 0.05) in HepG2 cells compared with the control group in a dose-dependent manner determined by MTT assay (Figure 1). YHK at 100 and 200 mg/L inhibited cell growth to 56% and 49% of the control, respectively, in HepG2 cells. A significant dose-response inhibition of incorporation of 3H-thymidine was observed by doses starting from 50 mg/L concentration (> 50% inhibition, p < 0.01) (data not shown).
Cell cytotoxicity assay
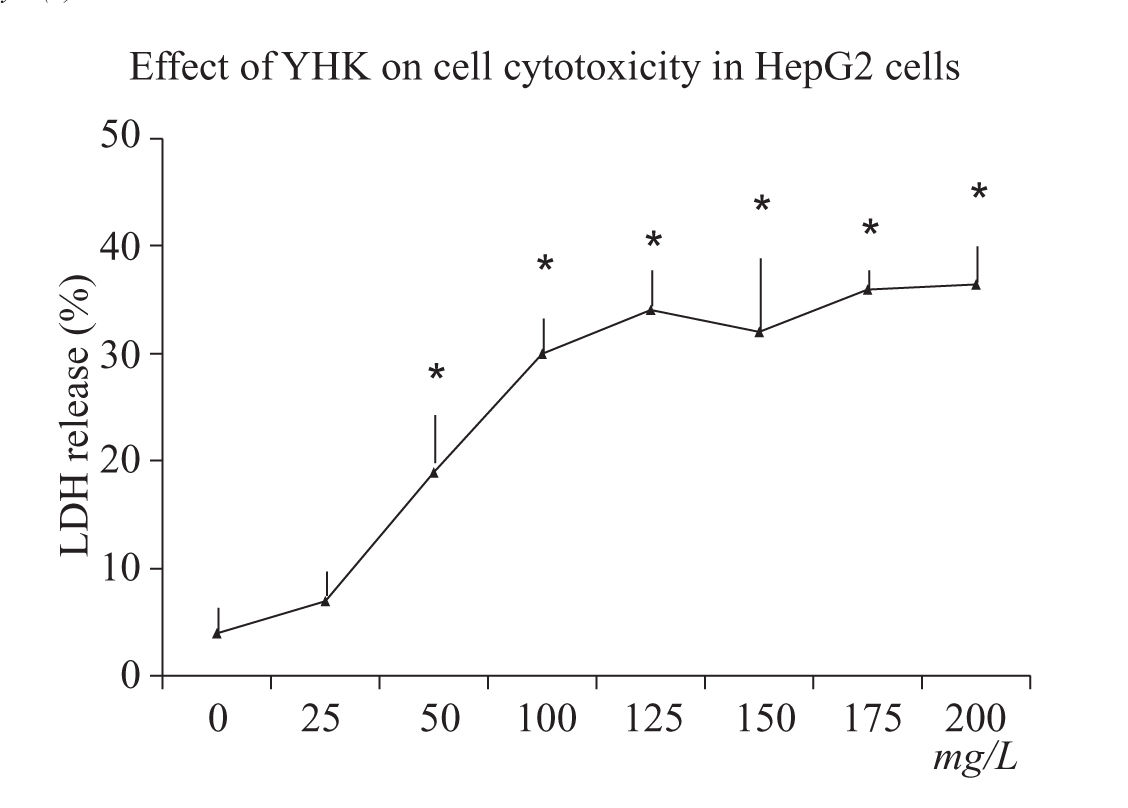
Cell cytotoxicity was directly measured by LDH release. After 24 h incubation, YHK (50-200 mg/L) significantly increased cell cytotoxicity (p < 0.05) in HepG2 cells compared with the control group (6.2%) (Figure 2). YHK at a dose of 200 mg/L significantly increased LDH release to 28.6% (p < 0.05) cells compared with the dose of 50 mg/L (12.2%).
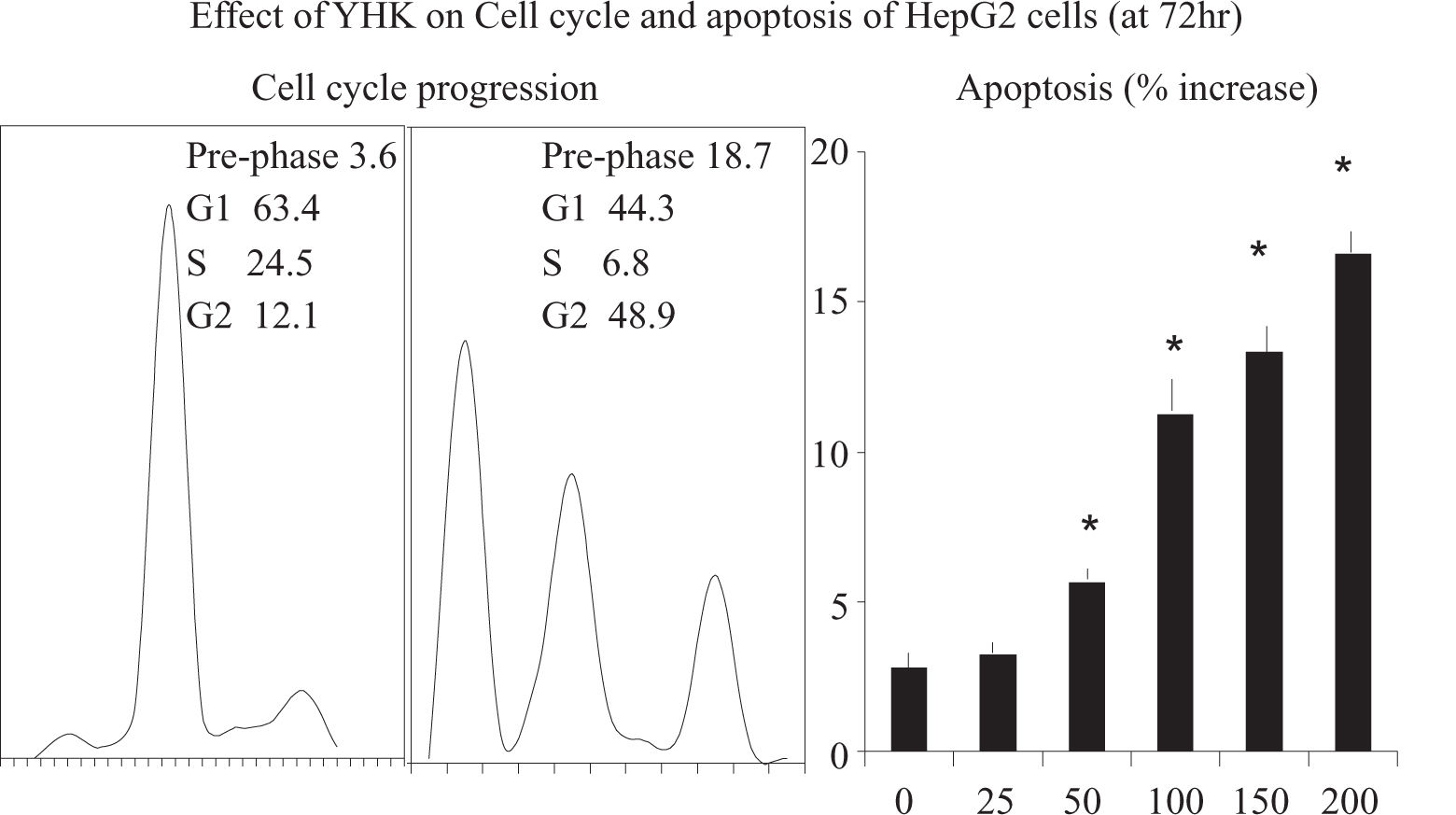
Effect of YHK on HepG2 cell cycle and apoptosis release. Data are expressed as x±s (n= 3). Values not sharing the same letter differed significantly (P < 0.05) in the same cell line by Fisher’s least significant difference test.")
The effect of YHK on cell cycle was assessed by flow cytometry. It appeared a dose-and time-dependent effect of YHK on the cell cycle of HepG2 cells. After 72 h of 200 mg/L YHK treatment, cells in the G2/M population increased from 12.1% to 48.9%, as compared to control. The increase of cell population at the G2/M phase was associated with a decrease of cell population in the G1 phase which dropped from 63.4% to 44.3% (p < 0.05, Figure 3). Accordingly, YHK induced a marked apoptosis at 72 h in a dose-dependent manner starting from 50 mg/L concentration. As expected, the increase in the apoptotic population was associated with a concomitant enlargement of the pre-phase population, as observed in the cell cycle analysis.
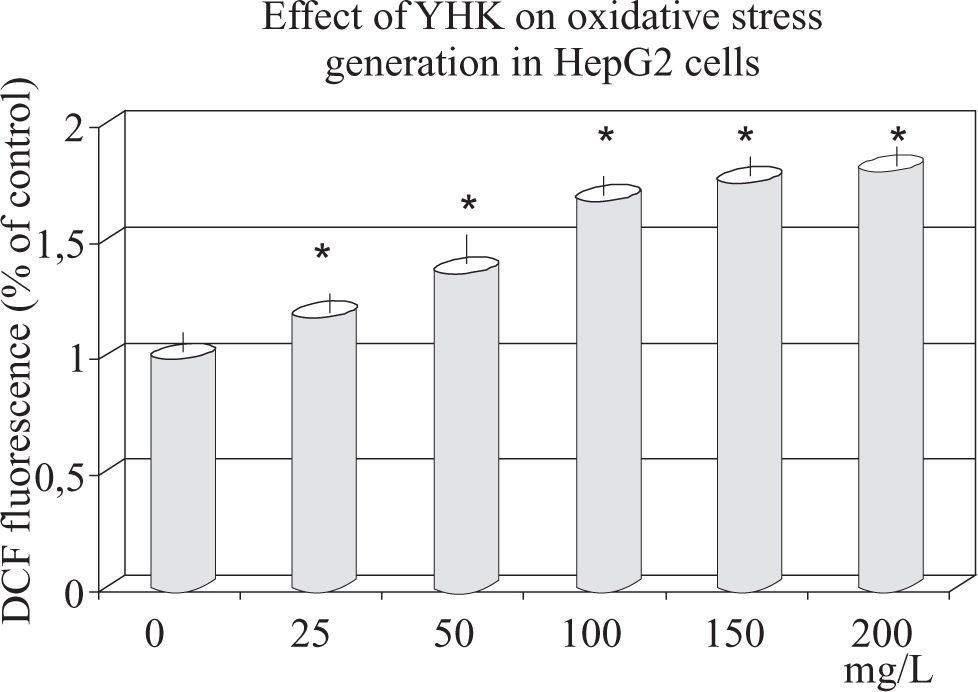
Effect of YHK on oxidative stress and related dose-related apoptosis enhancement as observed at 72 h. Data have been calculated over three measurements and are expressed as x±s.")
As shown in Figure 4, ROS level (corresponding to the fluorescence intensity) in HepG2 cells significantly increased after treatment with (50 mg/L to 200 mg/L) YHK. With highest concentration, ROS level increased up to 80% as compared to baseline (p < 0.001).
Discussion
Several experimental and epidemiological studies have shown that antioxidant-endowed micronutrients present in food can inhibit carcinogenesis by affecting the molecular events either in the initiation, promotion or progression phase.19-22 Apoptosis is an essential process for maintaining homeostasis in multi-cellular organisms and it is induced by receptor-regulatory signal such as hormones, cytokines and growth factors,23 thus playing crucial roles in a variety of physiological and pathological conditions. Indeed, anticancer drugs are expected to modulate the extrinsic and intrinsic regulators of apoptosis and sensitize cancer cells to apoptosis while, ideally, being devoid of cytotoxic effects on normal cells. In the present study, we observed that YHK can inhibit HepG2 cells growth in concentration-dependent manners and at 200 mg/L concentration the survival fraction decreased to nearly null (Figure 1). Besides the inhibitory effect of YHK on cell proliferation, high doses (100-200 mg/L) of YHK significantly increased cytotoxicity as determined by LDH release in HepG2 cells. However, the cytotoxic effect of YHK reached the plateau at the dose over 150 mg/L. The cytotoxic effect of YHK could be attributed to necrosis and/or apoptosis. Our result revealed increased LDH release as an index of necrosis in YHK-treated cells, indicating that YHK could trigger necrotic changes. There may exist the possibility that apoptosis contributes to the cytotoxic effect of YHK. Indeed, it has been shown that the induction of apoptosis is accompanied with the generation of intracellular ROS prior to the activation of caspase-324 and down-regulation of Bcl-2.25 Our results indicated that YHK either at apoptosis-inducing dose or not could cause significantly increased ROS levels as compared with control. It is well known that the ROS production either at intra and extra-cellular sites may influence the cell survival during oxidative insults26 while apoptosis may be regulated by the intracellular redox status.27 Interestingly, it has been recently shown that a natural compound Salvia miltiorrhiza, may inhibit human hepatoma HepG2 cells growth by inducing apoptosis28 bringing about also an intracellular GSH deletion, as shown in fundamental cellular studies.29 Accordingly, we suggest that YHK-induced high level of ROS may be one of the putative mechanisms involved in the apoptosis and in growth inhibition and of HepG2 cells, as reported by Simizu et al. for some anticancer drugs.30 YHK was shown to be able to induce a G2/M cell cycle arrest and a dose-and time-dependent apoptotic effect in HepG2 cells. Cell cycle checkpoints are important control mechanisms that ensure the proper performance of cell cycle events. In particular, the G2/M checkpoint blocks the entry into mitosis in the event DNA is damaged.31 In summary, the present study demonstrates that YHK has profound effects on HepG2 hepatocellular carcinoma cells in vitro. It reduces the proliferation of these cells and induces cell death by apoptosis. It leads to ROS generation and a likely redox imbalance in HepG2 cells, and it is speculated that the redox imbalance caused by YHK may play an important role in the growth inhibition and induction of apoptosis of HepG2 cells. The present study does not allow to discriminate each single component effect nor the possible interplay among them and this undoubtedly represent a limitation so far. However, at least some components contained in YHK, such as gallic acid and glycyrrhiza, have been shown to possess cancer growth inhibitory effect in similar in vitro and in vivo test.32-34 In particular, it has been demonstrated that gallic acid shows a selective intracellular ROS-related cytotoxicity against tumor cells with higher sensitivity than normal cells such as hepatocytes which are resistant probably due to the generation of the inhibitor against gallic acid-induced apoptosis.35 Our results not only provide the basis for further in vivo and clinical research in hepatocellular carcinoma, but also prompt further studies aimed to the understanding of the pharmacology of YHK in more details.



