



ATP-binding cassette (ABC) subfamily B member 4 (ABCB4), also known as multidrug resistance protein 3 (MDR3), encoded by ABCB4, is involved in biliary phospholipid secretion, protecting hepatobiliary system from deleterious detergent and lithogenic properties of the bile. ABCB4 mutations altering canalicular ABCB4 protein function and expression may have variable clinical presentation and predispose to several human liver diseases. Well-established phenotypes of ABCB4 deficit are: progressive familial intrahepatic cholestasis type 3, gallbladder disease 1 (syn. low phospholipid associated cholelithiasis syndrome), high ɣ-glutamyl transferase intrahepatic cholestasis of pregnancy, chronic cholangiopathy, and adult biliary fibrosis/cirrhosis. Moreover, ABCB4 aberrations may be involved in some cases of drug induced cholestasis, transient neonatal cholestasis, and parenteral nutrition-associated liver disease. Recently, genome-wide association studies have documented occurrence of malignant tumours, predominantly hepatobiliary malignancies, in patients with ABCB4/MDR3 deficit.
The patient's age at the time of the first presentation of cholestatic disease, as well as the severity of liver disorder and response to treatment are related to the ABCB4 allelic status. Mutational analysis of ABCB4 in patients and their families should be considered in all individuals with cholestasis of unknown aetiology, regardless of age and/or time of onset of the first symptoms.
Adenosine triphosphate (ATP)-binding cassette (ABC) subfamily B member 4 (ABCB4), also known as multidrug resistance protein 3 (MDR3 in humans, Mdr2 in mice), is the membrane-associated transport protein almost exclusively expressed in the liver [1,2]. ABCB4/MDR3 is encoded by ABCB4 gene (OMIM *171060), located on chromosome 7q21.1, consisting of 27 coding exons and spanning ∼74kb. The ABCB4 gene promoter harbours a CpG island that is usually unmethylated in normal cells [3,4].
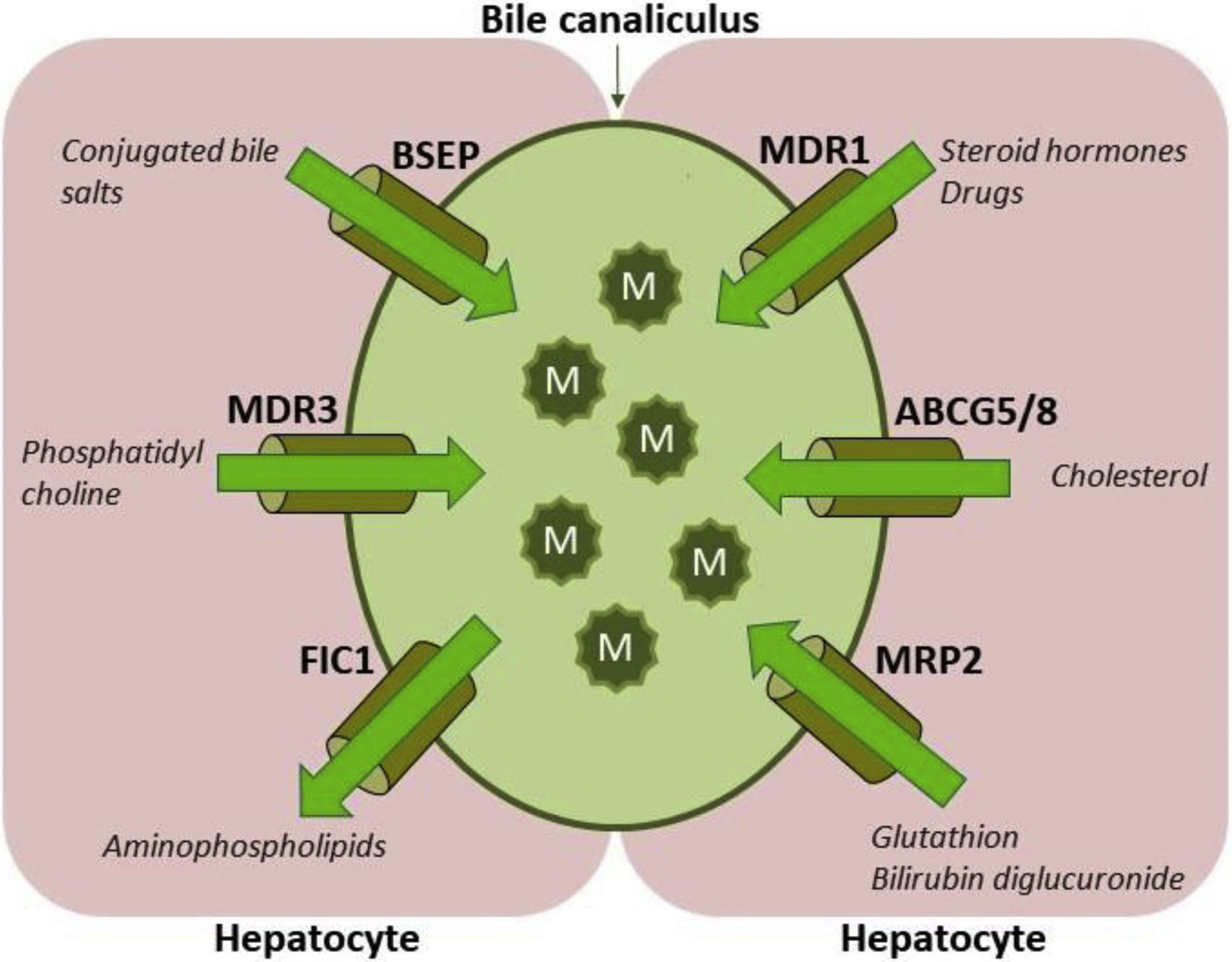
ABCB4/MDR3 P-glycoprotein consists of six intracellular domains and six extracellular loops separated by twelve transmembrane domains (TMDs). The protein contains two intracytoplasmic ATP-binding domains, also known as nucleotide binding domains (NBDs), with characteristic motifs Walker A, Walker B and ABC signature located upstream from the Walker B domain. The NBDs transfer energy to transmembrane transport of the substrate against concentration gradient, whilst the TMDs provide specificity for the substrate [1–3]. ABCB4/MDR3 is organised as full transporter and acts as an energy-dependent “floppase”, translocating phospholipids (PL) of the phosphatidylcholine (PC) family from the inner to the outer leaflet of the lipid bilayer of the canalicular membrane to be extracted by bile salts (BS). In primary bile, PC, BS and cholesterol form mixed micelles (Fig. 1). Disrupted formation of mixed micelles and accumulation of free nonmicellar BS results in production of bile with deleterious detergent properties to the membranes of cholangiocytes and hepatocytes. Additionally, biliary PL are involved in maintaining biliary cholesterol in solution, thus preventing its precipitation and subsequent formation of cholesterol crystals and stones [5–8].
![Hepatocyte canalicular transporters and bile formation. BSEP mediates the efflux of conjugated BS into bile. BS are essential for extraction of the membrane PC flopped by MDR3 from the inner to the outer leaflet of the membrane, as well as for the cholesterol efflux mediated by ABCG5/8 heterodimer. BS, PC and cholesterol form mixed micelles, reducing detergent and lithogenic activity of the bile. MRP2, a canalicular conjugate export pump, mediates excretion of a wide range of amphipathic anionic substrates, including bilirubin glucuronides and glutathione. MDR1 transports many metabolites and toxins of endogenous or exogenous origin into bile. FIC1 acts as aminophospholipid flippase, essential to maintain membrane lipid asymmetry responsible for canalicular membrane stability [2,6–8]. Legend: ABCG5/8: ATP-binding cassette, sub-family G, member 5/8, BS: bile salts, BSEP: Bile salt export pump, FIC1: Familial intrahepatic cholestasis, M: mixed micelles, MDR1: Multidrug resistance protein 1, MDR3: Multidrug resistance protein 3, MRP2: Multidrug resistance-associated protein 2, PC-phosphatidylcholine.](https://static.elsevier.es/multimedia/16652681/0000001900000002/v1_202003030740/S1665268119322628/v1_202003030740/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9DlRXbH51ovUyrMcgfU34eWVOJrGbFtgOUXqemEdRaQQPcCLkuecpOFm3dCzEWspNP+2pBPiNuH2acoP+urcfGUTSVn6gwSm3Pt5Czn6MurzVrkNmDm4BOZXNJt6iIEs6M6+FfABMhK9ty+KCFXzAjMfUXGu9ZNesJEqEBnSIPG86qydinAgr22J7C2vWgxMz9luxdJJrM5Wiu8akKKCrbljMKec8hTdFrxge/NfjPRJMw= "Hepatocyte canalicular transporters and bile formation. BSEP mediates the efflux of conjugated BS into bile. BS are essential for extraction of the membrane PC flopped by MDR3 from the inner to the outer leaflet of the membrane, as well as for the cholesterol efflux mediated by ABCG5/8 heterodimer. BS, PC and cholesterol form mixed micelles, reducing detergent and lithogenic activity of the bile. MRP2, a canalicular conjugate export pump, mediates excretion of a wide range of amphipathic anionic substrates, including bilirubin glucuronides and glutathione. MDR1 transports many metabolites and toxins of endogenous or exogenous origin into bile. FIC1 acts as aminophospholipid flippase, essential to maintain membrane lipid asymmetry responsible for canalicular membrane stability [2,6–8]. Legend: ABCG5/8: ATP-binding cassette, sub-family G, member 5/8, BS: bile salts, BSEP: Bile salt export pump, FIC1: Familial intrahepatic cholestasis, M: mixed micelles, MDR1: Multidrug resistance protein 1, MDR3: Multidrug resistance protein 3, MRP2: Multidrug resistance-associated protein 2, PC-phosphatidylcholine.")
Hepatocyte canalicular transporters and bile formation. BSEP mediates the efflux of conjugated BS into bile. BS are essential for extraction of the membrane PC flopped by MDR3 from the inner to the outer leaflet of the membrane, as well as for the cholesterol efflux mediated by ABCG5/8 heterodimer. BS, PC and cholesterol form mixed micelles, reducing detergent and lithogenic activity of the bile. MRP2, a canalicular conjugate export pump, mediates excretion of a wide range of amphipathic anionic substrates, including bilirubin glucuronides and glutathione. MDR1 transports many metabolites and toxins of endogenous or exogenous origin into bile. FIC1 acts as aminophospholipid flippase, essential to maintain membrane lipid asymmetry responsible for canalicular membrane stability [2,6–8]. Legend: ABCG5/8: ATP-binding cassette, sub-family G, member 5/8, BS: bile salts, BSEP: Bile salt export pump, FIC1: Familial intrahepatic cholestasis, M: mixed micelles, MDR1: Multidrug resistance protein 1, MDR3: Multidrug resistance protein 3, MRP2: Multidrug resistance-associated protein 2, PC-phosphatidylcholine.
Expression of ABCB4/MDR3 protein is restricted to the canalicular (apical) membrane of hepatocytes, but lower mRNA levels were also found in other normal tissues [3,4]. Study on foetal liver demonstrated that ABCB4 mRNA levels are 16-fold lower compared to the normal adult liver with only faint and focal canalicular MDR3 immunostaining, suggesting that ABCB4 develops late in gestation or even postnatally [7,9].
ABCB4 expression is regulated at several levels by transcriptional and posttranscriptional mechanisms. The nuclear farnesoid X receptor (FXR), a master regulator and coordinator of BS and lipid metabolism, seems to play an important role in ABCB4 regulations. ABCB4 is trans-activated by the FXR via a direct binding of FXR/retinoid X receptor α heterodimer to a highly conserved inverted repeat-1 (IR-1) motif at the distal promoter. Except BS-mediated induction of ABCB4 via FXR, fibrates upregulate ABCB4/MDR3 expression via the peroxisome proliferator-activated receptor α (PPARα) and facilitate hepatic export of PC [10–12].
PC has been recognised as endogenous ligand of the orphan nuclear receptor liver receptor homologue-1 (LRH-1, Lrh1 in mice, also NR5A2, gene NR5A2) [13,14]. The results of animal studies have demonstrated that Lrh1 is a transcriptional regulator of the floppase Mdr2 with corresponding effects on biliary PC secretion [15]. Interestingly, the transporter MDR3/Mdr2 might play an important role as modulator of glucose homeostasis and the effect could also be mediated by LRH-1-dependent PC pathways [14,16].
It has been demonstrated that ABCB4 allelic status correlates with phenotype and severity of liver disease [17,18]. According to the recently proposed functional classification, originally designed for PFIC3, variations in ABCB4 gene can be classified as nonsense mutations (class I), missense mutations affecting the maturation (class II), activity (class III), or stability of the ABCB4/MDR3 protein (class IV), and variations without identifiable effect on protein function and expression (class V) [19]. Approximately 300 disease causing ABCB4 variants have been reported, typically with homozygous status in rapidly progressive cholestatic liver disease with potentially life-threatening complications and with heterozygous status in less severe forms [19].
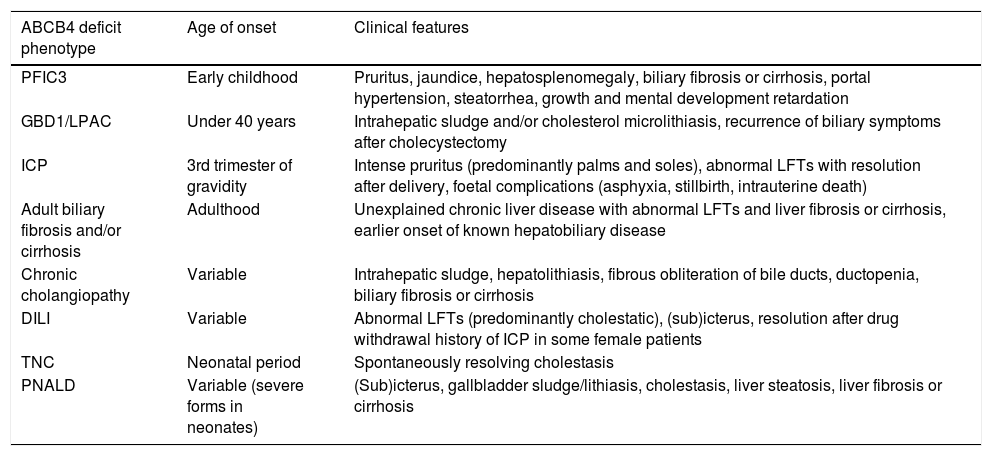
The main clinical spectrum of ABCB4 deficiency-associated diseases includes (Table 1): progressive familial intrahepatic cholestasis type 3 (PFIC 3), gallbladder disease 1 (GBD1), also known as low phospholipid-associated cholelithiasis syndrome (LPAC), intrahepatic cholestasis of pregnancy (ICP), adult biliary fibrosis or cirrhosis, chronic cholangiopathy, drug-induced liver injury (DILI), and transient neonatal cholestasis (TNC) [20–22]. Additionally, some variations in ABCB4 may be involved in the development of parenteral nutrition-associated liver disease (PNALD) [23,24]. Finally, the recent genome-wide association studies (GWAS), as well as family studies have found association between ABCB4 variants and primary hepatobiliary malignancies [25–27].
Phenotypic spectrum of ABCB4 disease.
| ABCB4 deficit phenotype | Age of onset | Clinical features |
|---|---|---|
| PFIC3 | Early childhood | Pruritus, jaundice, hepatosplenomegaly, biliary fibrosis or cirrhosis, portal hypertension, steatorrhea, growth and mental development retardation |
| GBD1/LPAC | Under 40 years | Intrahepatic sludge and/or cholesterol microlithiasis, recurrence of biliary symptoms after cholecystectomy |
| ICP | 3rd trimester of gravidity | Intense pruritus (predominantly palms and soles), abnormal LFTs with resolution after delivery, foetal complications (asphyxia, stillbirth, intrauterine death) |
| Adult biliary fibrosis and/or cirrhosis | Adulthood | Unexplained chronic liver disease with abnormal LFTs and liver fibrosis or cirrhosis, earlier onset of known hepatobiliary disease |
| Chronic cholangiopathy | Variable | Intrahepatic sludge, hepatolithiasis, fibrous obliteration of bile ducts, ductopenia, biliary fibrosis or cirrhosis |
| DILI | Variable | Abnormal LFTs (predominantly cholestatic), (sub)icterus, resolution after drug withdrawal history of ICP in some female patients |
| TNC | Neonatal period | Spontaneously resolving cholestasis |
| PNALD | Variable (severe forms in neonates) | (Sub)icterus, gallbladder sludge/lithiasis, cholestasis, liver steatosis, liver fibrosis or cirrhosis |
Legend: DILI: drug induced liver injury, GBD1/LPAC: gallbladder disease 1/low phospholipid-associated cholelithiasis, ICP: intrahepatic cholestasis of pregnancy, LFTs: liver function tests, PFIC3: progressive familial intrahepatic cholestasis type 3, PNALD: parenteral nutrition-associated liver disease, TNC: transient neonatal cholestasis.
Importantly, individuals with ABCB4/MDR3 deficiency might experience different clinical-pathological forms of the disease during their life, in some patients with overlapping phenotypes [22].
2Progressive familial intrahepatic cholestasis type 3Progressive familial intrahepatic cholestasis type 3 (PFIC3, OMIM #602347) is a life-threatening autosomal recessive cholestatic liver disorder caused by biallelic mutations in ABCB4, usually occurring during the first year of life or early childhood [28]. Clinically, PFIC3 is characterised by jaundice, severe itching, hepatosplenomegaly with signs of portal hypertension, steatorrhea, and retardation of growth and mental development. Malabsorption of fat-soluble vitamins may eventually result in episodes of repeated nosebleeds, abnormal susceptibility to bruising, and rickets, with progressive softening and weakness of the bones and predisposition to skeletal deformities and fractures. Extrahepatic manifestation of ABCB4/MDR3 deficit has not been reported [28]. Typical laboratory findings associated with PFIC3 are characterised by predominantly conjugated hyperbilirubinemia, increased levels of serum alkaline phosphatase (ALP) and serum alanine aminotransferase (ALT), and – in contrast to other PFIC types – by elevated serum ɣ-glutamyl transferase (GGT) activity. Serum concentrations of primary BS are increased, but concentrations of biliary BS remain within the normal range. Biliary lipid analysis shows decreased PL levels [28].
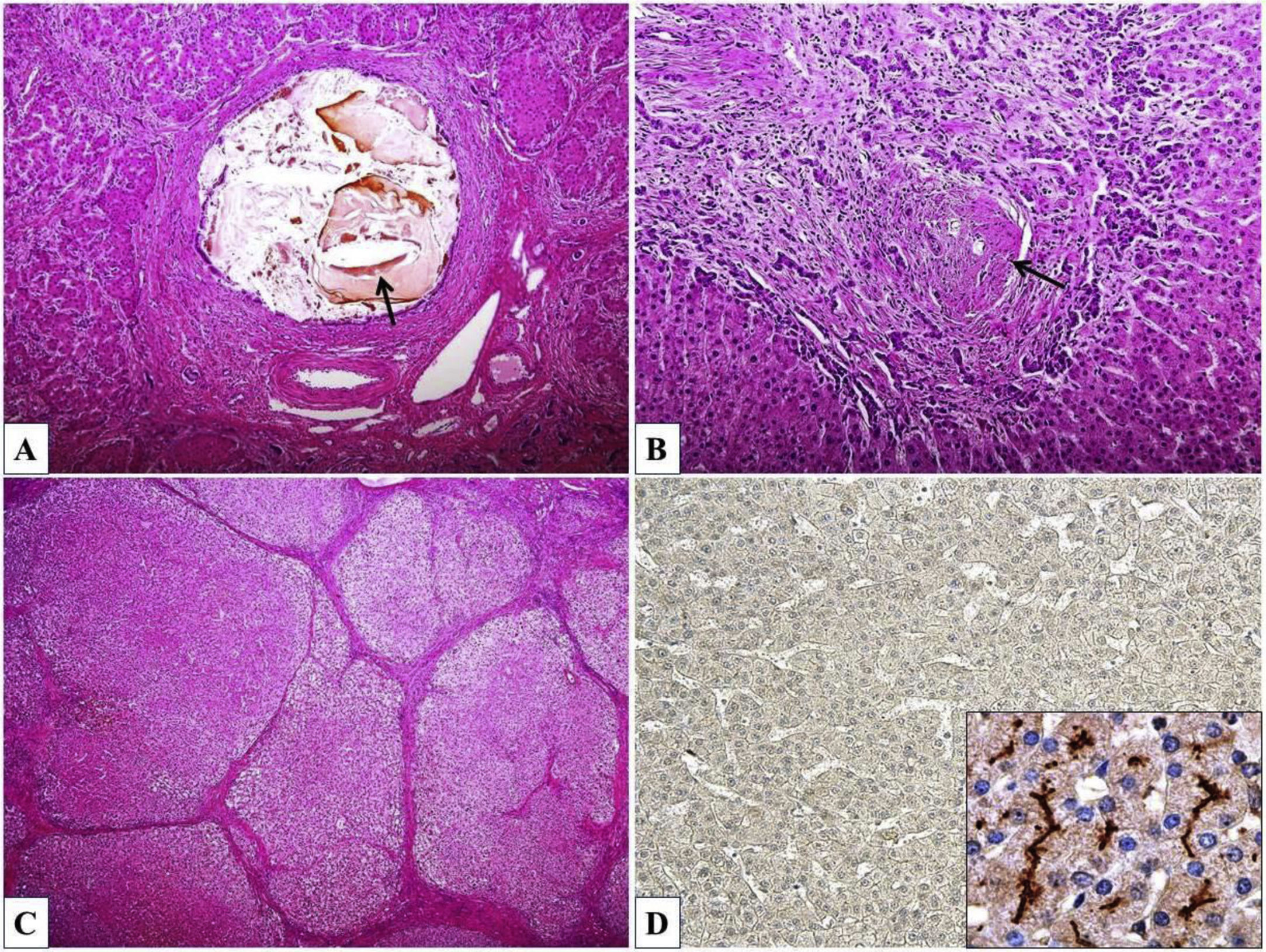
Histopathological findings associated with PFIC3 vary widely and include primarily portal-based inflammatory infiltrate with marked periportal ductular reaction accompanied by fibrosis, usually progressing to biliary-type cirrhosis, lobular disarray, giant-cell transformation of hepatocytes, and hepatocanalicular cholestasis. Cholesterol crystals within the bile ducts and morphological features of chronic cholangiopathy can be occasionally seen. Immunohistochemical examination of liver tissue demonstrates significant reduction or complete absence of canalicular MDR3 protein expression, predominantly in early onset forms (Fig. 2). Ultrastructurally, bile is dense and amorphous, occasionally with cholesterol clefts, in contrast to coarsely granular “Byler bile” in ATP8B1 deficiency (also known as PFIC1, OMIM #211600) [29].
 Inspissated bile and cholesterol cleft (arrow) within the dilated bile duct. (B) Chronic cholangiopathy with fibroobliterative portal bile duct lesion (arrow) and periportal ductular reaction. (C) Biliary cirrhosis with parenchymal nodules surrounded by fibrous septa. (D) Complete absence of ABCB4/MDR3 protein expression (inset – canalicular positivity in a healthy control). Haematoxylin & eosin, orig. magnif. ×100 (A), ×200 (B), ×40 (C); MDR3 immunohistochemistry (D), orig. magnif. ×100 (inset ×600).")
Histomorphology of ABCB4 deficiency. (A) Inspissated bile and cholesterol cleft (arrow) within the dilated bile duct. (B) Chronic cholangiopathy with fibroobliterative portal bile duct lesion (arrow) and periportal ductular reaction. (C) Biliary cirrhosis with parenchymal nodules surrounded by fibrous septa. (D) Complete absence of ABCB4/MDR3 protein expression (inset – canalicular positivity in a healthy control). Haematoxylin & eosin, orig. magnif. ×100 (A), ×200 (B), ×40 (C); MDR3 immunohistochemistry (D), orig. magnif. ×100 (inset ×600).
Without appropriate molecular genetic analysis, PFIC3 patients could be misdiagnosed as having other chronic cholestatic disease, hereditary or acquired, such as biliary atresia, Alagille syndrome or sclerosing cholangitis. Moreover, prolonged cholestasis in some PFIC3 patients may be accompanied by significant panlobular accumulation of copper in liver tissue, as well as by increased urine copper excretion. These findings may be potentially misinterpreted as Wilson's disease [18,30].
Effect of ursodeoxycholic acid (UDCA) treatment in PFIC3 patients is only intermediate, usually without complete normalisation of the liver function tests. More than half of PFIC3 cases progresses to end-stage liver disease and liver transplantation (LTX) is needed within the first decade of life [18,27].
3Gallbladder disease 1A subgroup of patients, predominantly young women, with ABCB4 variants, presents with gallbladder disease 1 (GBD1, OMIM #600803), also known as low phospholipid-associated cholelithiasis syndrome (LPAC), characterised by an increased risk for the early development of predominantly intrahepatic cholesterol gallstones [31]. Exact prevalence of the syndrome remains unknown. The GBD1 should be supposed in the patients with symptomatic cholelithiasis and at least one of the following criteria: age less than 40 years at onset of symptoms, recurrence after cholecystectomy, intrahepatic sludge or microlithiasis, and familial history of cholelithiasis in the first-degree relatives [31]. Interestingly, cholesterol lithiasis has also been demonstrated in patients with PFIC3 and their relatives, as well as and in the patients with a history of ICP [22].
Ultrasonography typically demonstrates intrahepatic hyperechogenic foci or hepatic lithiasis along the biliary tree. The cholangiographic findings in GBD1 include large unifocal or multifocal spindle-shaped dilations of the intrahepatic bile ducts [31,32]. The diagnosis of GBD1 can be confirmed by microscopic and chemical analysis of bile, typically containing aggregates of cholesterol crystals and/or small stones, and reduced levels of biliary PL. Molecular genetic analysis of ABCB4 should be used to confirm the diagnosis of GBD1/LPAC syndrome in young adults with symptomatic lithiasis and should allow familial screening [27,31].
Most of the patients with GBD1 benefits from long-term UDCA administration, either curative or prophylactic. Endoscopic retrograde cholangiography, biliary drainage and surgical treatment, i.e. cholecystectomy or liver resection, may be indicated in case of symptomatic lithiasis. Patients with end-stage liver disease are candidates for LTX [27,31].
4Intrahepatic cholestasis of pregnancyIntrahepatic cholestasis of pregnancy (ICP, OMIM #147480) is characterised by cholestasis and intense pruritus with onset in otherwise healthy pregnant women without a previous history of a liver disorder. It occurs in the third trimester and resolves completely soon after delivery [33,34]. Elevated levels of serum BS concentrations, predominantly serum cholic acid, and increased liver aminotransferase and ALP activity are typical laboratory findings. Although the disease is considered relatively benign for the mother, an increased rate of adverse foetal outcomes, including foetal asphyxia, stillbirth, or even intrauterine death, has been recorded [34].
In Europe, ICP affects about 1% of all pregnancies. Female sex hormones are considered to play an important role in pathogenesis of ICP, which is supported by the rapid cessation of cholestasis after delivery, the higher incidence of ICP in multiple pregnancies, and by the increased susceptibility of affected women to develop cholestasis under oral contraception [33,34]. However, the ethnic and geographical distribution of ICP indicates the importance of genetic predisposition to this condition. Except of mutations in ABCB11, ATP8B1, and nuclear receptor subfamily 1, group H, member 4 (NR1H4), heterozygous ABCB4 variations, predominantly in exon 14, have been identified in a group of ICP patients, specifically in those presenting with elevated serum GGT activity [35–37]. While GGT is normal in ABCB11-related forms or oestrogen-associated cholestasis, increased GGT levels could serve as discriminating factor for ABCB4 deficient cases.
Treatment with UDCA improves clinical symptoms in afflicted women, predominantly pruritus, and may even reduce the risk of adverse foetal outcome [38,39].
5Adult biliary fibrosis or cirrhosisThe link between heterozygous missense and nonsense ABCB4 mutations and unexplained liver fibrosis or biliary cirrhosis has been highlighted by several studies [21,25,40,41]. The large Icelandic GWAS demonstrated that ABCB4 variations not only represent the cause of rare monogenic liver diseases but also affect chronic liver disease in general [25]. The effect of common ABCB4 variant p.T175A as a potential modulator of liver stiffness was further supported by the study on two transient elastography-based cohorts of patients with chronic liver disorders [42].
Histomorphological phenotype associated with ABCB4 mutations includes portal fibrosis, in some patients with significant progression in the course of the disease, periportal ductular proliferation, and a predominantly macrophagic infiltration of the portal triads with no or only mild periportal or lobular necroinflammatory activity. Mild bile duct lesions have also been observed in some individuals [21].
An early diagnosis and prophylactic administration of UDCA may slow down fibrosis progression and delay a risk of potential complications, at least in some of the affected individuals [21,27].
6ABCB4 deficiency-associated cholangiopathyThe primary defect in ABCB4/MDR3 does not cause retention of BS in hepatocytes and the symptoms can be explained by the deleterious effect of detergent bile to the biliary tree [43,44]. More than two decades ago, an animal experiment has demonstrated an occurrence of nonsuppurative inflammatory bile duct lesions (cholangitis) in Abcb4−/− mice [45]. Similarly, cases of chronic cholangiopathy in humans, associated with ABCB4 variants, have been described [46–49]. Thus, ABCB4/MDR3 deficit represents more likely primary biliary lesion rather than primary hepatocellular cholestatic condition [44].
Morphological spectrum of bile duct lesions described in ABCB4 deficit varies widely. As mentioned above, patients with GBD1/LPAC syndrome present with intrahepatic spindle-shaped bile duct dilations, filled with inspissated bile or gallstones. The intrahepatic calculi occur mostly without morphological strictures of the intrahepatic bile ducts [31,32]. Light microscopy demonstrates intraductal aggregates of cholesterol crystals and small cholesterol stones in the GBD1 patients (Fig. 2) [29,31].
Whereas the main feature in Mdr2 knock-out mice is sclerosing cholangitis (SC), controversies exist whether a genetically determined alteration of ABCB4/MDR3 also plays a pathogenic role in primary biliary cholangitis and primary SC in humans [50]. However, MR imaging presentations mimicking SC corresponding to small duct fibro-obliterative lesions at light microscopical level have been reported in two patients with known ABCB4 deficit [32].
Several reports of liver cirrhosis accompanied with vanishing bile duct syndrome (ductopenia) in ABCB4/MDR3 deficient patients have also been published [47–49].
7Drug-induced liver injuryDrug-induced liver injury (DILI) is a form of acquired liver disease, presenting with a wide spectrum of acute of chronic clinical-pathological lesions, that could be classified as hepatocellular, cholestatic, and/or mixed types [51,52]. The functional and clinical impact of ABCB4 variations for the development of DILI has been still under investigation. However, several clinical studies have demonstrated that ABCB4 deficiency may predispose to predominantly cholestatic liver injury induced by oral-contraceptives, psychotropic drugs, selected chemotherapy drugs, statins, and antibiotics [53,54].
As mentioned above, contraceptive-induced liver injury has also been reported in women with a prior history of ICP [34].
Most cases of DILI resolve after withdrawal of the drug. Administration of UDCA may improve cholestasis induced by 17β-estradiol glucuronide [50]. Patients at risk for developing progressive liver disease, biliary cirrhosis, and/or liver failure should be referred to liver transplant centres.
8Transient neonatal cholestasisTransient neonatal cholestasis (TNC) is a form of spontaneously resolving cholestasis, probably multifactorial, with immaturity and perinatal distress, liver ischaemia or hypoxia being the most important offending factors [55,56]. However, in 10% of TNC cases no obvious aetiology can be identified and an underlying genetic defect in hepatobiliary transport system may be involved in the cholestatic process [55,56].
Several reports have documented an occurrence of TNC in ABCB4 deficient individuals [17,21,57]. A heterozygous mutation in ABCB4 may predispose to temporary decompensation of bile secretion processes, which are immature in neonates [21,55,57].
The management of TNC is predominantly supportive with particular attention to an adequate supplementation by fat-soluble vitamins. The use of UDCA is associated with improvement in biochemical parameters of cholestasis [56].
9Parenteral nutrition-associated liver diseaseParenteral nutrition-associated liver disease (PNALD) is defined by a direct bilirubin level of >2.0mg/dl following a prolonged course of total parenteral nutrition TPN (>2 weeks), if other possible causes of cholestasis have been ruled out [58]. Clinical and pathological manifestations of PNALD, such as steatosis, cholestasis, gallbladder sludge/stones, and liver fibrosis or cirrhosis, may occur separately or in combination [59].
The aetiology of PNALD is multifactorial, with prematurity, infection, history of bowel resection and/or lack of enteral feeds being of particular importance [58]. Neonates are more susceptible to severe forms of PNALD than older children or adults, that could be – at least partly – caused by late development of hepatobiliary transport systems, predominantly ABCB4/MDR3 [7,9]. Additionally, animal and human studies showed that the absence of enteral nutrition causes a generalised reduction of bile secretion, with a particular decrease of the Abcb4/ABCB4 function [60–62].
Recent studies have demonstrated, that the c.504 C>T and c.485 T>A ABCB4 mutations in exon 6, as well as the c.2793 frameshift mutation in exon 23 are possibly involved in the development of PNALD in premature infants [23,24].
10ABCB4 deficit-associated malignanciesThe data derived from two recent GWAS collected from Icelandic and Indian population identified risk ABCB4 variants associated with hepatobiliary malignancies, predominantly hepatocellular carcinoma (HCC), cholangiocarcinoma (CCA), and carcinoma of gallbladder [25,26,63].
HCC is the most frequent primary hepatocellular cancer in adult population and the major cause of liver-related death in patients with compensated cirrhosis [64]. The major risk factor of HCC is the presence of advanced fibrosis or cirrhosis caused by viral (chronic hepatitis B and C), toxic (alcohol and aflatoxins), metabolic (non-alcoholic fatty liver disease, hereditary haemochromatosis), and immune-related factors (primary biliary cirrhosis and autoimmune hepatitis) [65].
CCA, a malignant tumour originating from biliary epithelium, represents less than 5% of digestive neoplastic lesions. Congenital biliary abnormalities, PSC, and hepatolithiasis are independent risk factors for the development of either intra- or extrahepatic CCAs [66].
Gallbladder cancer is a relatively rare form of malignancy with dismal prognosis in most cases. The risk factors vary geographically and among ethnic groups, with chronic cholecystitis and gallstones being identified as the most important [67].
Interestingly, rare cases of paediatric HCC and CCA arising on the background of ABCB11 deficiency (PFIC2) have been reported in the literature, thus introducing the question of possible relationship between BS metabolism disturbance and carcinogenesis [68–70].
The underlying pathophysiological mechanisms of carcinogenesis in the setting of ABCB4 deficit in humans have yet to be elucidated. However, hypothesis of inflammation-driven carcinogenesis derived from the murine model of Mdr2 knock-out mice helps to shed light on complex processes of tumour occurrence and development [45,71]. Liver inflammation and toxicity induced by BS in Mdr2 deficient mice lead to the development of hepatocyte dysplasia, and by 16 months of age, to the occurrence of liver cancer in virtually 100% of animals [71]. Similarly, in humans, the inactive ABCB4/MDR3 transporter predisposes to BS-mediated hepatobiliary damage with necroinflammatory response, prolonged cholestasis, and subsequent fibrosis, which increases hepatocyte and cholangiocyte turnover, and growth of altered cells. All these events may ultimately lead to the development of neoplastic process [72,73].
Except of association between ABCB4 deficit and primary malignancies of liver parenchyma and biliary tree, recent studies have pointed to the importance of ABCB4/MDR3 transporter for non-hepatobiliary tumours. Epigenetic silencing of ABCB4, resp. Abcb4 through CpG island promoter hypermethylation, occurs in human, resp. murine tumours. Aberrant ABCB4 methylation was found in primary human lung cancer (39%), breast cancer (41%), and in head and neck cancer (20%) tissues. In 37% of primary lung cancer samples, ABCB4 expression was absent. Overexpression of ABCB4 significantly suppressed colony formation and proliferation of lung cancer cells [4]. ABCB4 mediates the efflux transport of doxorubicin and contributes to the acquired resistance of doxorubicin in breast cancer cells [74]. Additionally, ABCB4 may act as a tumour suppressor gene to regulate chemoresistance in colorectal cancer and knockdown of ABCB4 could predict a shorter recurrence-free survival and overall survival of the afflicted patients [75].
11ConclusionsABCB4 variants altering function of hepatocanalicular ABCB4/MDR3 transporter may predispose to a phenotypically heterogeneous spectrum of cholestatic liver disorders, typically manifested at the biochemical level by high activity of serum GGT and low concentration of PC in bile. Moreover, recent studies have demonstrated that some common ABCB4 variants may also act as potential modulators of liver fibrosis, both in primary cholestatic and primary non-cholestatic liver diseases, representing thus an important genetic determinant of chronic liver disorder progression and outcome [25,42]. The same large studies have also pointed to a possible link between alterations of ABCB4 and increased prevalence of hepatobiliary malignancies [25,26].
Most of the harmful effects of ABCB4 deficit on hepatobiliary system are attributable to “toxic bile” with potent detergent and lithogenic properties. Reduction of these deleterious effects of the bile can be achieved through supplementation with UDCA (3α,7β-dihydroxy-5β-cholan-24-oic acid), a secondary bile acid, first characterised in the bile of bears as a conjugate with taurine [76,77]. UDCA, more hydrophilic and less hepatotoxic compared to cholic and deoxycholic BS, is currently considered the first line therapy in ABCB4-deficient patients, in which promotes choleresis, competes with primary BS in the intestine, and suppresses de novo BS synthesis [77,78]. Except of changes in the hydrophobicity index of the BS pool, UDCA conjugates have immunomodulatory, anti-inflammatory and anti-apoptotic effects that may also contribute to cytoprotection of hepatocytes in cholestatic conditions [79–81]. More significant success by UDCA treatment can be achieved predominantly in the patients with milder phenotypes of the ABCB4 disease, in which some residual transport capacity is still preserved. Prophylactic or therapeutic UDCA administration may slow down progress to the end-stage liver disease and delay possible complications [80,81].
Intractable pruritus in PFIC3 patients could be alleviated by rifampicin, upregulating detoxification enzymes and export pumps. Moreover, serotonin antagonists and its reuptake inhibitors also seem to be beneficial in managing refractory pruritus in some PFIC3 patients [81,82]. Pharmacological chaperones (e.g. 4-phenylbutyrate), agonists of nuclear and membrane receptors (e.g., statins, fibrates, obeticholic acid), cystic fibrosis transmembrane conductance regulator potentiator (ivacaftor – VX-770), and/or endoplasmic reticulum-associated degradation inhibitors (MG132) can increase the expression of functional proteins and potentially mitigate the ABCB4 deficient phenotypes, predominantly in combination with UDCA [80–83].
Despite the indisputable benefit of conservative therapeutic methods, LTX has still remained the ultimate treatment modality for some ABCB4 deficient patients, predominantly those with rapidly progressive liver disease and/or with complications resistant to conservative therapy [18,84]. Interestingly, serious post-transplant complications known from PFIC1 and PFIC2, such as worsening of diarrhoea, an occurrence of steatohepatitis, and/or recurrence of PFIC phenotype in the liver graft have not been reported in PFIC3 patients so far [85].
Molecular analysis of ABCB4 in probands and their families is beneficial for prenatal diagnosis, prevention and treatment of afflicted individuals and should be considered in all patients with cholestasis of unknown aetiology, regardless of age and/or the time of onset of the first symptoms. The recently introduced functional classification of ABCB4 variations on the basis of molecular defect helps not only to understand a correlation between genotype and phenotype of the disease, but also provides a useful tool for future individualised mutation-targeted therapeutic strategies [19].AbbreviationsABC ATP-binding cassette subfamily ATP-binding cassette, subfamily B, member 4 ATP-binding cassette, subfamily B, member 11 ATP-binding cassette, sub-family G, member 5/8 alkaline phosphatase alanine aminotransferase bile salts bile salt export pump cholangiocarcinoma drug induced liver disease familial intrahepatic cholestasis farnesoid X-activated receptor gallbladder disease 1 ɣ-glutamyl transferase genome-wide association study hepatocellular carcinoma intrahepatic cholestasis of pregnancy inverted repeat DNA element with 1 nucleotide spacing low phospholipid-associated cholelithiasis syndrome liver receptor homologue 1 liver allograft transplantation multidrug resistance protein 1 multidrug resistance protein 2 (mouse) multidrug resistance protein 3 nucleotide binding domain nuclear receptor subfamily 1, group H, member 4 electronic database Online Mendelian Inheritance in ManTM phosphatidylcholine progressive familial intrahepatic cholestasis phospholipid parenteral nutrition associated liver disease peroxisome proliferator-activated receptor α retinoid X receptor sclerosing cholangitis transient neonatal cholestasis transmembrane domain ursodeoxycholic acid
This study was funded by the Ministry of Health of the Czech Republic [grant number NV18-06-00032].
Conflict of interestThe authors have no conflicts of interest to declare.



