



Lipoproteins are synthesized by the liver and secreted to plasma. Chronic alcoholic intoxication produces frequently cirrhosis and concomitantly alterations in liver metabolism. Thirty patients with alcoholic cirrhosis and 83 healthy controls were selected for this study. Apolipoprotein A1, B100, lecithin cholesterol acyltransferase, responsible for cholesterol esterification and seudocholinesterase enzyme activity not related to lipid metabolism, as a referent of proteins synthesized by the liver were analyzed. In 7 patients serum tiobarbituric acids, catalase, glutathione peroxidase were measured, as exponent of the presence of oxidative stress. Our results showed a significant decrease in lipoproteins, lecithin cholesterol acyltransferase and seudocholinesterase activities. An increase in serum tiobarbituric acids and a decrease in both antioxidant enzymes were found as well. In conclusion, alcohol cirrhotic liver decreases the production of liver proteins including those related to lipid metabolism, allowing the formation of steatosis and/or necrosis. Moreover oxidative stress participate possible as a major mechanism in liver damage.
Lipoproteins are synthesized by the liver and then secreted to plasma.1-3 Different kinds of pharmacological and toxic agents can modify their structure and disturb its metabolism.3-5
Prolonged ingestion of alcoholic beverages, carbon tetrachloride intoxication, hepatotoxic drugs, such as cisplatinun, tamoxifene, bupremorphine among others, as well as metabolic insulin resistance syndrome, can produce alterations in lipid liver metabolism inducing liver steatosis and/or necrosis.6 In liver fatty infiltration Virus C aggression can also be present, as well as in Reye´s syndrome.7-12
Under certain pathological conditions, as occur in chronic ethanol exposure, reactive oxygen species (ROS) production is increased and the level of antioxidant substances and enzymes are reduced. This imbalance between ROS production and its removal constitutes the process called oxidative stress (OS).13 Almost all these noxious agents previously referred, produce alterations on liver lipid metabolism, and other tissues. They act not only on lipid metabolism, but basically on liver proteins, glycophospholipids, ceramides including an important number of enzymes that participate in its metabolism producing finally liver injury.14,15
It is known that chronic alcoholic liver disease develops because the presence of alcohol and its metabolites damage its parenchymal and non-parenchymal hepatic cells.16 Furthermore, alcohol produces its toxic effect on the intestinal wall, allowing among other changes, the passage of bacterial toxins to the splanchnic blood flow, reaching the liver through the portal circulation.
Chronic alcohol ingestion generates besides, reactive oxygen species (ROS) production, cytokines and immune reactions, constituting an alcoholic systemic disease.17-18 When the production of ROS overwhelms the liver antioxidant capacity of liver cells, the damage of cellular macromolecules involved in liver metabolic pathways are deranged in humans and rat models.19
Mitochondria respiratory chain generate most of the ROS produced in the body. Liver is one of the major source of ROS, through cytochrome P-450 mixed function oxidases.20
Besides alcohol has been shown to deplete glutathione GSH levels, particularly in the mitochondria, which normally possess high levels of imported GSH from the cytosol by a carrier protein. This GSH is incharge partially of ROS neutralization, that is formed during the respiratory chain activity. Alcohol seems to interfere with this protein carrier in this step of GSH passage from cytosol to mitochondria.21
In liver, the mitochondrial transport of GSH is also regulated by carriers and membrane fluidity. Alcohol ingestion that produces liver damage, induces a decrease in this membrane fluidity, worsening the GSH transport.21-23
Besides it results probable that in alcoholic cirrhotics, the lack of adequate nutrients, decrease the levels of some aminoacids, as is the case of cysteine, that constitutes a part of the glutathione molecule and its defect weaken the antioxidant system, by producing less glutathione.
The aim of this study was to determine the apolipoprotein AI, B100, the cholesterol esterifying lecithin-cholesterol acyl transferase (LCAT), the liver enzyme seudocholinesterase (CHE), «as a non lipid protein parameter» in chronic alcoholic patients. Tiobarbiturate reactive substances (TBARS), catalse (CAT), glutatione peroxidase (GSH-PX) were determined in a small group of patients as representative of an oxidative stress condition.
MethodsPatientsThirty alcoholic cirrhotic patients and 83 control subjects were selected from the Gastrointestinal and Clinical Unit of a General Hospital of the City of Buenos Aires (Pirovano I. Hospital) Argentina.
All the patients included in this study, presented a prolonged (more than ten years) an important daily alcoholic intake, with clinical and biochemical signs of liver injury. Twenty-six of the patients were male and four female, with an average of 50 years old (range 25-65); the Child-Pugh classification was applied.24 The patients were divided in to two groups: G-1, with a compensate clinical state (n = 10): Child Pugh A; and G-2 with a decompesated state (n = 14): Child B and C, according with their clinical and biochemical situation; (50% of these patients were hospitalized). No hemorrhage was detected in these patients in the last four months before their inclusion in this study.
The 83 control subjects do not present hepatic, toxic or metabolic diseases. Sixty of them were males and twenty-three female, with the average age of 50 years old (range: 40-60).
Eighty percent of the alcoholic patients were submitted to a percutaneous liver biopsy, for pathological analysis, according to the presence of acceptable coagulation parameters. Abdominal ultrasonography examination was applied to detect hepatic-splenic morphology and to assess the presence and amount of peritoneal fluid. Complete clinical and Rx examinations was performed in all the patients.
The project of this work was approved of the Ethics Committee of the Faculty of Pharmacy and Biochemistry and the Hospital Investigation Committee.
Laboratory methodsComplete hematological tests, serum protein electrophoresis and coagulation tests were made. It was also determined the serum activities of alanino aminotransferase (ALT), aspartate aminotransferase (AST), gamma glutamyl transpeptidase (GGT), alkaline phosphatase (FAL), creatine kinase (CK) and seudocholinesterase (CHE). All the enzymes activities were measured by standardized and optimized commercial kits. Electrophoretic lipidograms on agarose gel was done (not included in this paper). Cholesterol, HDL-cholesterol and triglycerides were also determined by conventional methods.
Serum apolipoproteins, B100 and AI, were determined by electroinmunodifussion (SEBIA, France). Serum lecithin cholesterol acyl transferase (LCAT) was studied by the method of endogenous substrate, measuring changes in the relation: free cholesterol/esterified cholesterol, after serum incubation 3 hs at 37 °C.
Hepatic A, B, C viruses were tested with corresponding antibodies reaction.
Protein determinationPlasma protein concentration was evaluated by the method of Lowry et al,25 using bovine serum albumin as standard.
Antioxidant enzyme assaysCatalase (CAT) and glutathione peroxidase (GSH-Px) activities were determined spectrophotometrically in serum prepared in a medium consisting of 140 mM KCl and 25 mM potassium phosphate buffer (pH 7.4), and centrifuged at 600 x g for 10 min. CAT activity was determined by measuring the decrease in absorbance at 240 nm,22 GSH-Px activity following NADPH oxidation at 340 nm.26
Lipid peroxidation.Lipid peroxidation in serum was determined by measuring the rate of production of tiobarbituric acid reactive substances (TBARS), expressed as malondialdehyde equivalents.27,28 One volume of serum was mixed with 0.5 volume trichlororoacetic acid (15% w/v) and centrifuged at 2000 x g for 10 min. The supernatant (1 mL) was mixed with 0.5 mL thiobarbituric acid (0.7 g/L) and boiled for 10 min. After cooling, sample absorbance was read spectrophotometrically at 535 nm. Malondialdehyde concentration was calculated using a ϵ value of 1.56 x 105 M-1cm-1.
Statistical analysisIt was applied t-Student’s test for independence samples. Differences were considered significant with a p < 0.05.
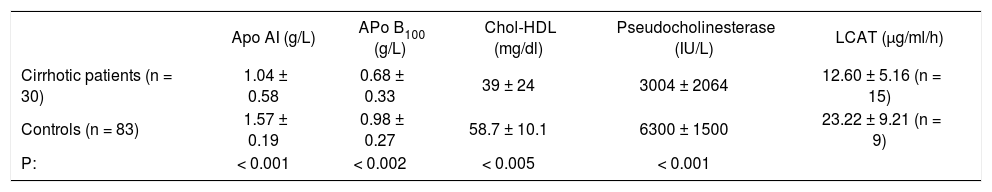
ResultsThe determination of both apolipoproteins measured in cirrhotic patients serum was compared against control healthy subjects. As can be seen in table 1 the 30 alcoholic cirrhotic showed a decrease of serum Apo AI concentration of: 1.04 ± 0.58 g/L; Apo B 100: 0.85 ± 0.33 g/L, as compared to controls (Table I). The 83 control subjects showed the following values: Apo AI: 1.57 ± 0.19 g/L; Apo B 100: 0.98 ± 0.27 g/L, with a significance of p < 0.001 and p < 0.002 respectively between both groups.
Serum concentrations of apolipoproteins AI and B100, cholesterol HDL, pseudocholinesterase and lecitin cholesterolasyltransferase.
| Apo AI (g/L) | APo B100 (g/L) | Chol-HDL (mg/dl) | Pseudocholinesterase (IU/L) | LCAT (μg/ml/h) | |
|---|---|---|---|---|---|
| Cirrhotic patients (n = 30) | 1.04 ± 0.58 | 0.68 ± 0.33 | 39 ± 24 | 3004 ± 2064 | 12.60 ± 5.16 (n = 15) |
| Controls (n = 83) | 1.57 ± 0.19 | 0.98 ± 0.27 | 58.7 ± 10.1 | 6300 ± 1500 | 23.22 ± 9.21 (n = 9) |
| P: | < 0.001 | < 0.002 | < 0.005 | < 0.001 |
Key: Apo AI. Apolipoproteins AI; Apo B100, Apolipoproteins B100; Chol-HDL, cholesterol HDL; LCAT, lecitin cholesterolasyltransferase.
In the 30 cirrhotics patients, HDL-cholesterol values also decreased 39.07 ± 24.0 mg/dl against controls: 58.7 ± 10.1 mg/dl, the significance was p < 0.005. The activity of the serum enzyme CHE (not related to lipid metabolism) was 3004 ± 2004 UI/L as an average in 30 cirrhotics, against 6300 ± 1500 UI/L (Table II) in control patients with a p < 0.001.
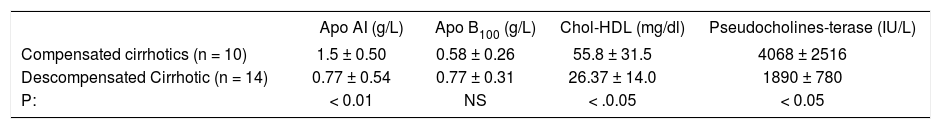
Differences between compensated and decompensated alcoholic cirrhotic patients.
| Apo AI (g/L) | Apo B100 (g/L) | Chol-HDL (mg/dl) | Pseudocholines-terase (IU/L) | |
|---|---|---|---|---|
| Compensated cirrhotics (n = 10) | 1.5 ± 0.50 | 0.58 ± 0.26 | 55.8 ± 31.5 | 4068 ± 2516 |
| Descompensated Cirrhotic (n = 14) | 0.77 ± 0.54 | 0.77 ± 0.31 | 26.37 ± 14.0 | 1890 ± 780 |
| P: | < 0.01 | NS | < .0.05 | < 0.05 |
Key: Apo AI, Apolipoproteins; Apo B100, Apolipoproteins B100; Chol-HDL, Cholesterol HDL.
Cirrhotic patients were divided in compensated, Child Pugh A (n = 10) with almost normal lipid serum profiles and descompensated patients, Child Pugh B and C (n =14). The results demonstrated that this second group showed a significant decrease in the activity of CHE against the first (p < 0.05), coincidently with the decrease in Apo AI concentration (p < 0.01) and HDL-cholesterol (P < 0.05), Apo B 100 did not show significant differences between both groups.
LCAT activity in cirrhotics decreased significantly, as compared to controls with a p < 0.028 (Table III).
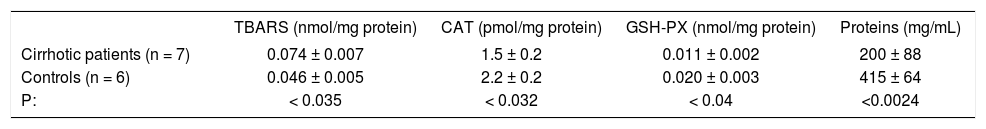
Oxidative stress parameters in the compensated cirrhotic patients.
| TBARS (nmol/mg protein) | CAT (pmol/mg protein) | GSH-PX (nmol/mg protein) | Proteins (mg/mL) | |
|---|---|---|---|---|
| Cirrhotic patients (n = 7) | 0.074 ± 0.007 | 1.5 ± 0.2 | 0.011 ± 0.002 | 200 ± 88 |
| Controls (n = 6) | 0.046 ± 0.005 | 2.2 ± 0.2 | 0.020 ± 0.003 | 415 ± 64 |
| P: | < 0.035 | < 0.032 | < 0.04 | <0.0024 |
Key: TBARS, Thiobarbituric acid reactive species; CAT, Catalase; GSH-PX, glutathione peroxidase.
In some of our decompensated cirrhotic patients and in 5 controls subjects, we measured in serum the following determinations: TBARS, CAT, GSH-Px, finding a statistically significant increase in TBARS (p < 0.035); and a decrease also significant of CAT, GSH-PX and proteins (p < 0.032, p < 0.04 and p < 0.0024 respectively) in the alcoholic patients (Table III).
In the last group of patients the number of serum tests diminished because some of them abandoned the hospital or died due to cirrhotic complications.
DiscussionThe metabolism of lipids involves four basic compartments: the hepatic parenchyma, the systemic blood flow, the adipose tissue and the intestine. The central nervous system (CNS), particularly the autonomic nervous system, and the inner secretion glands, participate also in this metabolism.
In this report that includes 30 cirrhotic alcoholics patients and 83 healthy control subjects, a significant decrease in serum Apo A1 and Apo B100, same as HDL-cholesterol and serum LCAT values, were found and compared to serum of control patients. The serum enzyme CHE activity, not related to lipid metabolism, and produced basically by the liver, also decreased significantly in cirrhotics, suggesting that these decreases can include almost all the proteins produced by the liver. Decreases in concentration were more marked in decompensated patients.
The hepatocyte is the major site of lipoprotein synthesis, its interconnection with other molecules and its release to plasma. Lipoproteins contain mainly triglycerides that join to their protein, apolipoproteins, as in the case of Apo AI, B100 and C in the endoplasmic reticulum, and then released through Golgi structure, out of the hepatocyte. Very low-density lipoproteins (VLDL) contain a relatively small amount of cholesterol and phospholipids, derived from «de novo» synthesis. The hepatocyte receives lipids from the plasma compartment, mainly in the form of low-density lipoproteins (LDL). Both lipoproteins, VLDL and LDL, contain a relatively high amount of cholesterol-ester and are, therefore, key particles in cholesterol homeostasis.
The phospholipid molecules play a fundamental role in the process of lipoproteins secretion from liver to circulation, nevertheless in this experiment phospholipid were not determined because it was emphasized the study basically on the protein defects in liver from alcoholic cirrhotics.
In the regulation of lipid synthesis and secretion, the presence of phosphatidylcholine is involved in the transport of apolipoprotein B and triacylglycerol.
According to Mathur et al experiments on isolated CaCo-2 cells, normal behavior of secretory pathways needs basically the phosphatidylcholine presence as to increase triacylglycerol-rich lipoprotein secretion.29 Utilizing Hep G2 cells, Zhou et al30 suggested that physiologic concentration of lysophosphatidylcholine modulates hepatic apo B secretion.
The circulating lipoprotein lipase enzyme in plasma participates in the production of LDL from VLDL, undergoing extensive modification. The nascent high density lipoprotein (HDL) particles are synthesized by the liver and intestine, and acquire free cholesterol from the plasma membrane cells at the peripheral tissues. Free cholesterol is converted into cholesterol ester by the action of a hepatic formed enzyme LCAT, and the mechanism of esterification occurs into the circulating HDL, inducing to form its spherical shape.
A reverse transport of cholesterol is produced when HDL is subsequently delivered back to the liver or transformed into LDL by the action of the cholesterol ester transfer protein.
Besides the formation of the lipoprotein Apo AI, B100 and LCAT, the liver synthesizes Apo C, Apo CIII, Apo E. Recently, according to Acton et al,31 a scavenger receptor B 1 (SR-B1) responsible for part of the HDL formation, was identified. This receptor was localized in other sites, where cholesterol HDL uptake occurred, as is the case of the ovaries, testis, adrenals and the liver as well. The SR-B1 receptor apparently regulated the level of biliary cholesterol.32 Beside, HDL constitutes a special source of biliary lipids.33,34
Among other proteins normally produced by the liver, and related to lipid metabolism can be mentioned, the hepatic lipase,35 that hydrolyzes the lipoproteins of intermediary density (IDL), the paroxonase associated to HDL, the farnesoid X receptor, related to Apo CIII.36 The cholesterol ester transport protein and the nuclear receptors activators of peroxisomal proliferation (PPAR) were PPAR• • is the most common, also contributed to lipid metabolism.37-40
We include in this investigation the measure of the activity of the serum enzyme CHE, synthesized mainly by the liver, with the purpose to use it as a non-lipid protein to compare its plasma concentration to lipid metabolism related proteins.
It is noteworthy recall that LCAT, utilize HDL as a normal substrate.39-44 This enzyme transfers a fatty acid from lecithin to free cholesterol forming a cholesterol ester. The decreased activity of plasma LCAT in cirrhotic patients, increases the amount of free fatty acid (FFA) concentration and is associated to FFA uptake defects by the damaged liver.
FFA are taken up by the normal liver, and then are esterified to triglyceride, and in a lesser extent to phospholipid, diglyceride and cholesterol ester, or oxidized to CO2 and ketone bodies.
FFA circulate in blood attached to albumin, and in chronic liver injury lesser amounts of albumin is formed and commonly, these patients are undernourished, FFA increase in concentration as in a moderate starvation and triglycerides associated to lipoproteins are not secreted normally by the liver. In this situation, FFA-triglycerides accumulate in liver producing hepatic steatosis, and ketogenesis is accelerated as well. Besides, FFA can stimulate cholesterogenesis and also can be inhibited by cholesterol in VLDL remnants LDL and HDL. According to these facts, it can be suggested that in cirrhotic patients, FFA metabolism can be distorted dramatically with repercussion on total lipid metabolism. It must be recalled that free fatty acids and the long-chain acylcarnitine as well, have detergent properties at high concentration, on membranes, including those of mitochondria, and can inhibit Na+ K+ ATPase, with consequences on energy disposal.
We have mentioned an important number of liver proteins related to lipid metabolism, that can be special targets to toxic substances, as is the case of alcohol that disturbs its complex mechanism.
It also must be remembered that CNS constitutes also a major target in chronic liver alcohol intoxication, but apparently some of these patients, can present serious brain damage, but a moderate liver injury as demonstrated by liver biopsy.
Prolonged liver intoxication by alcohol produce liver damage utilizing two mechanisms, first during the alcohol metabolism by the direct toxic action of alcohol and its principal metabolite, acetaldehyde, acting on parenchymal and non-parenchymal cells (Kupfer cells, endothelial and stellate liver cells), and secondary by the formation of adducts and interleukins, that act as an additional effect to the first mechanism.13,16,17
Depending of the period of intake and amount of alcohol ingested it is produced a subtractive effect by the functional and morphological alteration of hepatic tissue, by fibrosis, necrosis and (or) inflammation.
Alcohol breakdown in liver parenchyma, result in the formation of molecules that lead to the ROS production and its increase in serum as demonstrated in a group of our patients. Alcohol stimulates the activity of cytocrome P-450, group of oxidizing enzymes contributing to the ROS formation.45
In the presence of alcohol ingestion, the liver is forced to use an increasing amount of oxygen, including that used in alcohol metabolism. This induces to oxygen deficits in some cortical areas of the cells, essential for cell survival, as in the reaction of glycolysis and mitochondrial energy process. The energy production alteration generated by alcohol is another factor that enhance liver cells damage.
Mitochondria respiratory chain, form most of the ROS produced in the body. Liver is a major sources of ROS basically by the CyP450 mixed function oxidases.20Studies of Hirano et al23 Zhao et al46 and Fernandez-Checa et al47 using a rat model of alcohol intoxication, demonstrated that chronic alcohol intoxication reduced the levels of mGSH in hepatocites that can be attributed to a defective transport of GSH from cytosol to mitochondria,48 and to a decreased in its membrane fluidity.
In cirrhosis, mitochondrial dysfunction is present,49and alterations in mGSH are found, similar to those found in bile duct legated rats, with depletion of mGSH.
In prior studies we demonstrate that bile duct ligation with extrahepatic cholestasis in rats, produce in liver severe disturbances on microsomal membrane structures and metabolic pathway including membrane-bound enzymes, critically dependent on the intact phospholipid environment, as phosphatidylcholine, phosphatidylserine, phosphatidylglycerol, among others.50 It must be recalled that in liver cirrhosis some intrahepatic cholestasis usually can be present.
The depletion of mGSH has been shown to sensitize cell to cytokine (TNF) or some drug-induce cell apoptosis.
Actually, according to Mari et al51 free cholesterol, but not free fatty acids or tryglicerides, sensitizes to TNF and Fas-induced steatohepatitis. Free cholesterol loading in mitochondria is apparently responsible for the hepatocyite sensitivity to TNF due to mitochondrial glutathione depletion.
In seven of our patients, serum TBARS, CAT, GSH-Px were measured, showing a significant increase in TBARS concentration and a marked decrease in the two antioxidant enzyme concentration, showing the existence of an oxidative stress condition.
In liver parenchyma the presence of metals, as in the case of free iron and zinc, enhances in cirrhotic livers the production of more oxygen radicals.44,45,52 Aldehyde formation associated to lipids and proteins, form certain adducts, that participated in liver injury.
The simultaneous presence in our alcoholic patients of a decrease in plasma apolipoproteins, LCAT, CHE and a significant increase in plasma TBARS and the decrease in CAT and GSH-PX as well, suggest the important role of OS, among other factors during alcohol intoxication and can be assumed its participation in its proteins and lipid derangements.
In conclusion, chronic liver disease produced by prolonged alcoholic ingestion as occurred in our 30 patients, produced serious impairments in lipid-protein liver metabolism. This was documented observing a significant decrease in serum of both apolipoproteins and LCAT activity as well. These findings suggest a marked alteration in these proteins synthesis and on its posterior blood transport to the periphery. The fall in activity of the serum CHE, synthesized basically in the liver suggests general toxic mechanism, that targeted almost all liver proteins damaging the clinical and biochemical condition of the patients.
The presence in a small group of decompensated alcoholic cirrhotic patients with increases of OS parameters confirm as described by recent papers, that OS has an important negative role in the mechanism of lipid liver metabolism and producing liver damage.



