




Background and rationale for the study. IL-17, TGF-β1/2 are cytokines involved in the development of kidney, pulmonary and liver fibrosis. However, their expression kinetics in the pathogenesis of cholestatic liver fibrosis have not yet been fully explored. The aim of the study was to analyze the expression of IL-17, RORγt, NKp46, TGF-β1, and TGF-β2 in the liver of rats with bile duct ligation (BDL).
Results. Hepatic IL-17A gene expression analyzed by qRT-PCR showed a dramatic increase of 350 and 10 fold, at 8 and 30 days post BDL, respectively. TGFβ1 and TGFβ2 gene expression significantly increased throughout the whole fibrotic process. At the protein level in liver homogenates, IL-17, TGF-β1, and RORγt significantly increased at 8 and 30 days after BDL. Interestingly, a significant increase in the protein levels of TGF-β2 and decrease of NKp46 was observed only 30 days after BDL. Unexpectedly, TGF-β2 exhibited stronger signals than TGF-β1 at the gene expression and protein levels. Histological analysis showed bile duct proliferation and collagen deposition.
Conclusions. Our results suggest that pro-fibrogenic cytokines IL-17, TGF-β1 and, strikingly, TGF-β2 might be important players of liver damage in the pathogenesis of early and advanced experimental cholestatic fibrosis. Th17 cells might represent an important source of IL-17, while NK cell depletion may account for the perpetuation of liver damage in the BDL model.
Liver fibrosis may result from a variety of chronic liver diseases, including hepatitis B, hepatitis C, and alcoholic liver disease. It is characterized by the accumulation of extracellular matrix (ECM) and scar formation. Several injury-triggered events playing critical roles in the pathogenesis of liver fibrosis, such as recruitment of inflammatory cells to the damaged area and profibrogenic cytokines release, have been described. However, transforming growth factor beta (TGF-β) and resident hepatic stellate cells (HSC) constitute the major mediators of liver injury.1,2
TGF-β is a member of a large family of multifunctional cytokines that exist in at least three isoforms denominated β1, β2, and β3, presenting 80% homology in their amino acid sequence. TGF-β1 binds to specific receptors on the cell surface, transducing signals to the nucleus by means of Smad proteins, upregulating collagen gene expression and resulting in collagen deposition within the liver.3 TGF-β1 isoforms play distinct roles in wound healing with TGF-β1/2 having predominantly pro-scarring roles and TGF-β3 having mainly anti-scarring effects.4–6 TGF-β2 transcripts have been identified in the bile duct epitelial cells of patients with biliary atresia, alcoholic cirrhosis, and cirrhosis secondary to hepatitis B.7,8 The participation of this cytokine in remodeling the ECM that stimulates the canonical Smad signaling pathway through Smad 2/3 in the pathogenesis of extraocular muscles fibrosis has been reported by Wordinger, et al.9 To date, reports dealing with TGF-β2 kinetics in the progression of chronic hepatic injury involved in cholestatic processes in humans and experimental models are limited.
Recently, the profibrogenic role of interleukin 17 (IL-17), a new cytokine, has been proposed in skin, lung, and liver fibrosis.10–12 IL-17A is the founding member of the IL-17 cytokine family, which now includes six major isoforms, IL-17A, B, C, D, E, and F. IL-17F is most closely related to IL-17A. The two molecules share 50% amino acid sequence homology. Different reports have demonstrated that IL-17A and IL-17F share some biological activities including the induction of cytokines and chemokines involved in inflammatory responses.13,14 However, additional activities specifically concerning to each cytokine have not been fully elucidated.
Association studies in patients with chronic liver injury by alcohol, hepatitis B, and hepatitis C show overexpression of IL-17, which interestingly correlates with the degree of fibrosis.15,16 Experimentally, dramatic decrease of liver injury and fibrosis in IL-17A or IL-17R(-/-) knockout mice with induced cholestatic or hepatotoxic fibrosis has been reported.17,18 On the other hand, IL-17 represents the most important cytokine produced by Th17 cells. Differentation of Th17 cells requieres the combined action of IL-6, IL-1β, TGF-β, and IL-23. These cytokines promote the expression of the lineage-specific transcription factor orphan nuclear receptor γt (ROR yt: in mice) or RORc (in human), wich is necessary and sufficient for development of Th17 cells.18,19 Therefore, RORγt would suggest the presence of Th17 cells.
The production of IL-17 could not be restricted only to Th17 cells, but other cell populations, such as CD8+T cells, γδT cells, and natural killer (NK) cells, may be a source for this cytokine. Considering the fact that up to 30% of the lymphocytes in hepatic parenchyma are represented by NK cells, it is factible to speculate that activated NK cells might be additional producers of IL-17. Several activating receptors, such as NKp46 and NKG2D, characterize to these cells in rodent models; however only the first one is mostly restricted to NK cells.20
Notwithstanding this, IL-17 expression in the different stages of the bile duct ligation (BDL)-induced cholestatic fibrosis, as well as its source, is unknown.
Based on this background, it is important to elucidate the IL-17, TGF-β1, and TGF-β2 expression kinetic pattern in the progress of experimental fibrosis, as well as the participation of Th17 and NK cells as major IL-17 sources.
Material and MethodsCholestasis-induced by bile duct ligationMale Wistar rats weighing 200-250 g, obtained from Charles Rivers (Boston, MA, USA) used in this study were kept under 12:12 h light/dark cycles, room temperature of 22 °C, constant humidity, and free access to food and water. Cholestatic fibrosis was induced by bile duct ligation (BDL) according to the method of Lee, et al., 1986.21 Animals were anesthetized with 30 mg/kg i.p. of Zoletil 50 (Virbac). Laparotomy was performed under antiseptic conditions. A mid-line incision in the abdomen exposing the muscle layers and the linea alba was made. The edge of the liver was then raised and the duodenum pulled down to expose the common bile duct. The common bile duct was doubly ligated with 4-0 silk thread and sectioned between the ligatures. Incisions were closed with silk thread. Control rats (sham) underwent the same surgical procedure except for ligation and section of the bile duct. Ligated animals (n = 15) were divided into two experimental groups (BDL), one corresponding to 8 days and the other one to 30 days evolution BDL, respectively. Finally, 5 survivor animals remained in each BDL group; meanwhile all sham animals (control) survived (n = 10). Experimental and control animals were sacrificed on day 8 or on day 30 post-surgery and representative liver samples were obtained.
All animal procedures were performed according to the guidelines of the Ethics Committee of University of Guadalajara based on technical specifications for the production, care, and use of laboratory animals NOM-062-ZOO-1999 and the International Guiding Principles for Biomedical Research Involving Animals established in 1985.22,23
RNA extraction and cDNA synthesisIsolation of total RNA from rat livers was carried out according to the method described by Chomczynski and Sacchi.24 Briefly, liver tissue was obtained from three different lobes and homogenized utilizing a Polytron system in the presence of Trizol (Invitrogen, Carlsbad, CA, USA). Two of RNA were reverse-transcribed in 0.05 M Tris-HCl, pH 8.3, 40 mM KCl, 7 mM MgCl2 buffer containing 0.05 µg/µL random hexamers, 1 mM dNTP mix, 0.05 U/mL RNAse inhibitor, and 200 U/mL M-MLV reverse transcriptase. Samples were incubated for 10 min at 70 °C, and prior to incubation for 60 min at 37 °C. Reverse transcriptase was inactivated by heating the sample tubes to 95 °C for 10 min, and the complementary DNA (cDNA) obtained were used immediately or stored at -20 °C until use.
Quantitative-reverse transcriptase-polymerase chain reactionThe expression of IL-17A, TGF-βΙ, and TGF-β2 genes was analyzed by quantitative-reverse transcriptase-polymerase chain reaction (qRT-PCR). We employed Oligo 7 primer analysis software (Molecular Biology Insights, Inc., USA) to design primers from NCBI GenBank Nucleotide database sequences. Primers were synthesized by integrated DNA Technologies (USA). Primer sequences, amplicon length, and aligning temperature are shown in table 1. All qRT-PCR reactions were performed in a final volume of 15 consisting of 3 of 1X PCR buffer (Light cycler fast start DNA master plus reaction mix SYBRgreen I, Roche Applied Science, Basel, Switzerland), 0.5 μΜ of sense and antisense primers, and 100 ng of cDNA. Reaction conditions were the following: 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and at 60 °C for 30 s. Additionally, melting analysis was performed, setting reactions at 95 °C for 10 sec, at 60 °C for 1 min, and by slowly heating the samples to 95 °C at a temperature rate of 0.1 °C/s. TaqMan probes (Applied Biosystems, Foster City, CA, USA) to amplify TGF-β2 were employed. Gene amplification was normalized against 18S rRNA expression. Complementary DNA (cDNA) was analyzed by triplicate and mean values were calculated. Relative quantification by the 2-ΔΔCT method was carried out, considering control groups as an internal calibrator.
Primer set sequences for quantitative-Reverse transcriptase-polymerase chain reaction (qRT-PCR) assays.
| Gene | Bases (n) | Sequence | Product length | Tm °C | |
|---|---|---|---|---|---|
| TGF-β1 Rat | F | 21 | TCCTTGCCCTCTACAACCAAC | 115 | 60 |
| R | 20 | TCCACCTTGGGCTTGCGACC | |||
| IL-17A Rat | F | 21 | CCGAGATAACTTTGAGGCATA | 292 | 62 |
| R | 20 | AACGAGGTTTGACTTTCACA | |||
| TGF-β2 Rat | Gene Bank Acces NM_031131 I.D | 95 | 60 | ||
| Rn00579674_m1 (applied Biosystem) |
Detection of TGF-β1, TGF-β2, IL-17, ROR-γ, and NKp46 was performed by Western blotting. Proteins were extracted from liver homogenates using RIPA buffer (SIGMA R 0278; Sigma, NJ, USA). Proteases inhibitor cocktail (Santa Cruz Biotechnology SC-29030, Santa Cruz, CA, USA) was added and clarified by centrifugation at 4 °C 12,000 rpm for 15 min. Protein concentration was determined by the Bradford method (Coomassie brilliant blue G-250, 95% ethanol, concentrated phosphoric acid and water). The total protein (40 g) was mixed with loading buffer under reducing conditions, electrophoresed on 7.5 -12% SDS-polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Bio-Rad, CA, USA). Non-specific binding was blocked with 3% milk. Subsequently, membranes were incubated with primary antibodies overnight: TGF-β1 (1:500) (sc - 52893), TGF-β2 (1:500) (sc - 374659), IL-17 (1:500) (sc - 374218), ROR-γt (1:500) (sc - 28559), and NKp46 (1:500) (sc - 292746). All antibodies were from Santa Cruz Biotechnology. HRP-conjugated anti-mouse or anti-rabbit secondary antibodies were used to reveal the blots: anti-mouse IgG (1:5,000) (SC - 2005) and anti-rabbit IgG (1:5000) (SC - 2004) were from Santa Cruz Biotechnology. Immune detection was performed using a chemiluminescence imaging system (Millipore, Billerica, MA, USA). Densitometric analysis was carried out with the Kodak MI 5.0 Image Analyzer (Kodak, Rochester, NY, USA). As an internal control to confirm that similar amounts of protein were loaded into each lane, GAPDH levels were determined using a monoclonal anti-gapdh IgG (1:5,000) (sc - 32233) and were revealed with anti-mouse IgG peroxidase (sc-2005).
Histological analysisLiver sections were randomly taken from the right, median, and left lobes of rat livers in each experimental group and immediately fixed by immersion in 4% paraformaldehyde diluted in phosphate saline buffer. The tissue were dehydrated in graded ethylic alcohol, and embedded in paraffin. Sections 5 μm thick were stained with Masson’s trichrome and analysed by light microscopy for determination of pathological changes.
Statistical methodsStatistical analysis was performed with SPSS Statistics software ver. 20.0 (SPSS, Inc., Chicago, IL, USA). Normally distributed variables were analyzed using one-way Analysis of variance (ANOVA) or the Student t test. Bonferroni and Dunnett post-hoc tests were employed for determination of statistical significance. Data are shown as the mean ± standard deviation (SD) of the mean. Only p values ≤ 0.05 were considered significant.
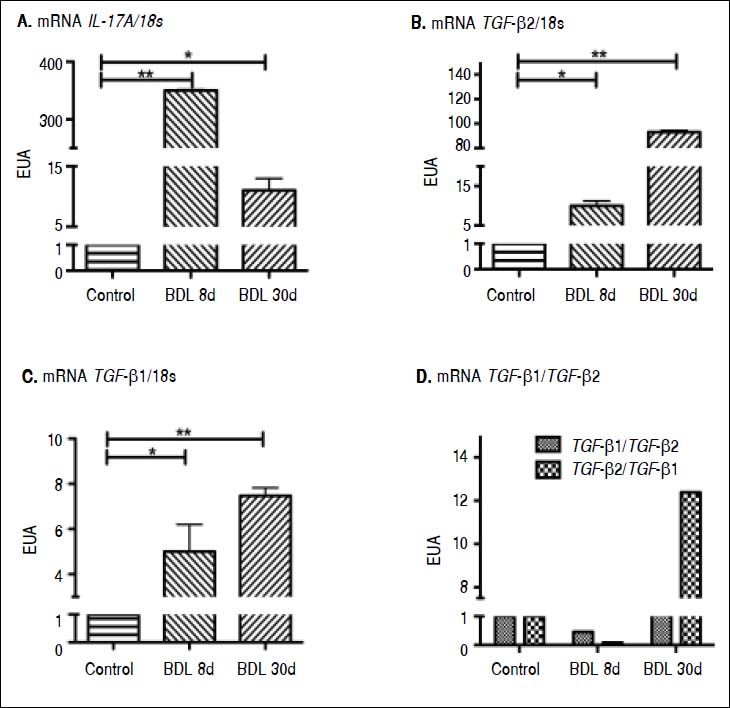
ResultsCytokine expression in the liver tissue was determined by qRT-PCR. A highly significant increase in IL-17A expression of 350 (p < 0.001) and 10 times (p < 0.05) more induction than the control after 8 and 30 days of BDL respectively, was observed (Figure 1A).
 method and ribosomal 18s gene expression as housekeeping gene. Results are expressed as Expression relative units (ERU). Each experiment was performed in triplicate. A. IL-17A expression, *p ≤ 0.05 control vs. BDL day 30, **p ≤ 0.001 control vs. BDL day 8. B. TGF-β2 expression, * p ≤ 0.05 control vs. BDL day 8, ** p ≤ 0.001 control vs. BDL day 30. C. TGF-β 1 expression, * p ≤ 0.05 control vs. BDL day 8; **p ≤ 0.001 control vs. BDL day 30, and (D) TGF-β1TTGF-β2 ratio EUA.")
Gene expression of IL-17A, TGF-β1, and TGF-β2 by qRT-PCR. The relative quantification was carried out utilizing the 2-ΔΔCt ± standard deviation (SD) method and ribosomal 18s gene expression as housekeeping gene. Results are expressed as Expression relative units (ERU). Each experiment was performed in triplicate. A. IL-17A expression, *p ≤ 0.05 control vs. BDL day 30, **p ≤ 0.001 control vs. BDL day 8. B. TGF-β2 expression, * p ≤ 0.05 control vs. BDL day 8, ** p ≤ 0.001 control vs. BDL day 30. C. TGF-β 1 expression, * p ≤ 0.05 control vs. BDL day 8; **p ≤ 0.001 control vs. BDL day 30, and (D) TGF-β1TTGF-β2 ratio EUA.
A very interesting behavior was exhibited by TGF-β2, which showed a significant increase up to 10 times more than that exhibited by the control group (p < 0.05), during early stages of fibrosis. More importantly, TGF-β2 was strikingly overexpressed (up to 93 times more) at 30 days after BDL when compared to the control group (p < 0.001), as seen in figure 1B.
Regarding TGF-β1 expression, it was induced 5 times (p < 0.05) and 7.5 times (p < 0.001) after 8 and 30 days BDL, respectively, when compared with control (Figure 1C). It is noteworthy to mention that a prevailing expression pattern of TGF-β2 over TGF-β1 of 2:1 after 8 and 30 days BDL was appreciated (Figure 1D).
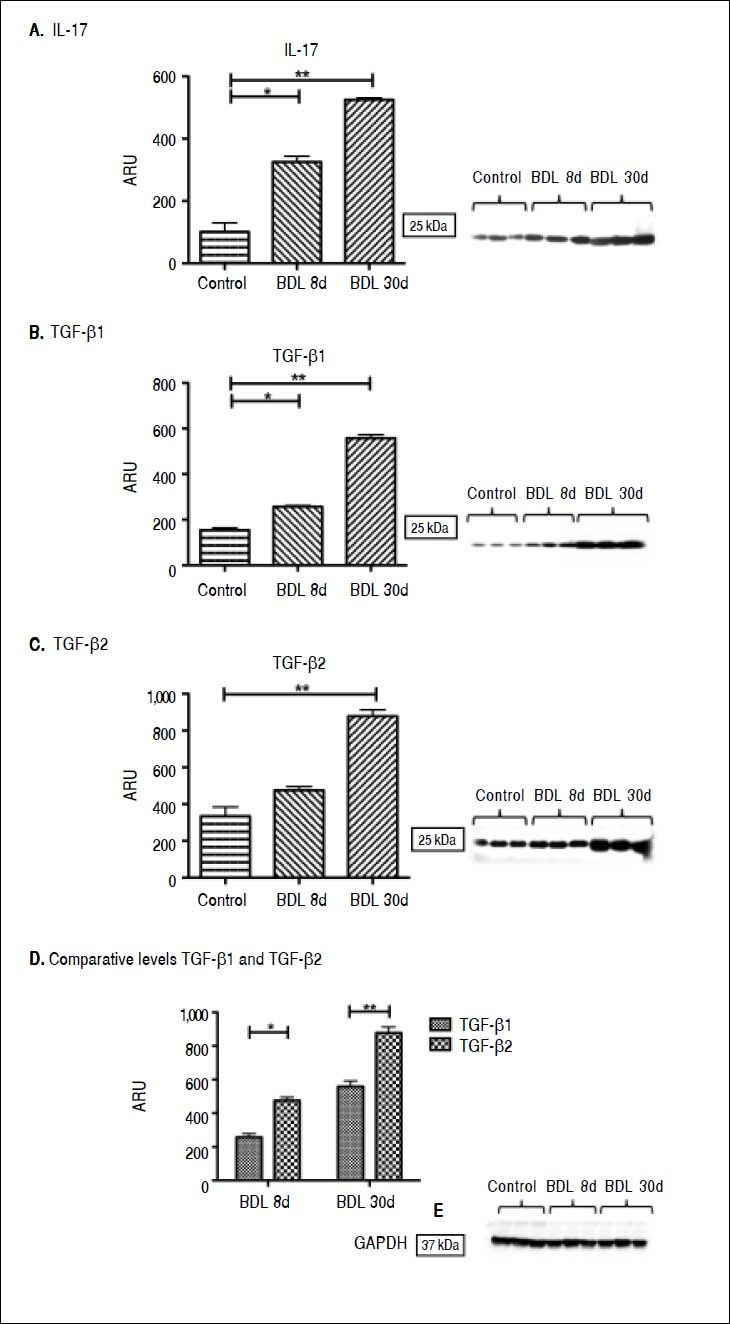
At the protein level in the liver tissue, significant IL-17 elevation of 2 and 3.5 times more than the control after 8 (p < 0.05) and 30 days (p < 0.001) BDL was exhibited (Figure 2A). However, the strongest effect was appreciated at the gene expression level.
 of five rats. Results are expressed as area relative units (ARU). A. IL-17 protein, 25 kDa, * p ≤ 0.05 control vs. BDL day 8; **p ≤ 0.001 control vs. BDL day 30. B. TGF-β 1 protein, 25 kDa, * p ≤ 0.05 control vs. BDL day 8; **p ≤ 0.001 control vs. BDL day 30. C. TGF-β2 protein, 25 kDa, ** p ≤ 0.001 control vs. BDL day 30. D. Comparative levels TGF-β1 and TGF-β2 proteins, * p ≤ 0.05 BDL day 8, * p ≤ 0.001 BDL day 30. E. GAPDH protein, 37 kDa.")
IL-17, TGF-β 1, and TGF-β2 proteins determined by Western blot. GA-PDH was used as an internal control. Each bar represents the mean value ± standard deviation (SD) of five rats. Results are expressed as area relative units (ARU). A. IL-17 protein, 25 kDa, * p ≤ 0.05 control vs. BDL day 8; **p ≤ 0.001 control vs. BDL day 30. B. TGF-β 1 protein, 25 kDa, * p ≤ 0.05 control vs. BDL day 8; **p ≤ 0.001 control vs. BDL day 30. C. TGF-β2 protein, 25 kDa, ** p ≤ 0.001 control vs. BDL day 30. D. Comparative levels TGF-β1 and TGF-β2 proteins, * p ≤ 0.05 BDL day 8, * p ≤ 0.001 BDL day 30. E. GAPDH protein, 37 kDa.
A significant increase in TGF-β1 concentration after 8 (257 ± 5 ARU vs. 154 ± 10 ARU; p < 0.05) and 30 days (555 ± 15 ARU vs. 154 ± 10 ARU; p < 0.001) BDL in comparison with control was observed (Figure 2B). Likewise, TGF-β2 showed a tendency toward increasing after 8 days BDL, and a significant increase (877 ± 35 ARU vs. 334 ± 50 ARU; p < 0.001) in the late stages of fibrosis when compared to control (Figure 2C). A prevailing pattern of TGF-β2 over TGF-β1 after 8 (p < 0.05) and 30 days (p < 0.001) BDL at the protein level similar to that already described at the expression level, was exhibited (Figure 2D).
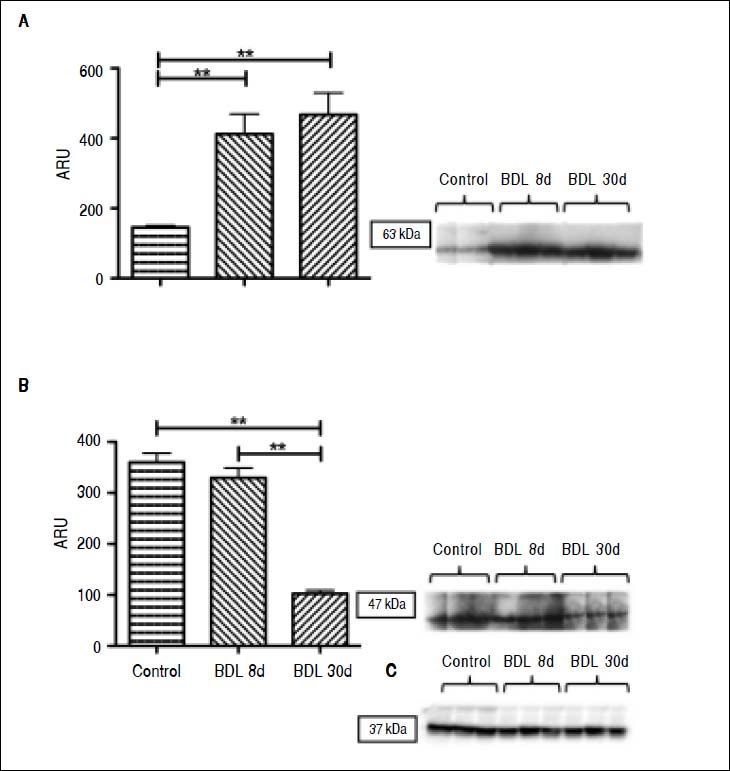
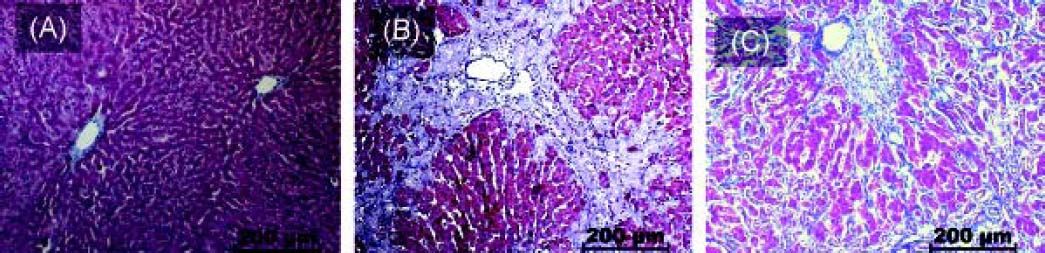
Interestingly, there was an increase of three times more than the control of ROR-γt (p < 0.001), a Th17 cell-specific transcriptional factor, throughout the whole fibrotic process (Figure 3A). In contrast, liver NKp46 level was significantly (p < 0.001) reduced after 30 days BDL when compared with the control (Figure 3B). Additionally, histopathological analysis exhibited normal morphology of the portal triad and hepatic lobules in control animals (Figure 4A); meanwhile, duct proliferation and periductal fibrosis after 8 days BDL, could be appreciated. This pattern became more apparent and substituting for nearly all of the hepatic parenchyma after 30 days BDL (Figures 4B and 4C).
 of five rats. Results are expressed as area relative units (ARU). A. ROR-γt protein, 63 kDa; ** p ≤ 0.001 control vs. BDL day 8, ** p ≤ 0.001 control vs. BDL day 30. B. NKp46 protein, 47 kDa, ** p ≤ 0.001 control vs. BDL day 30, ** p ≤ 0.001 BDL day 8 vs. BDL day 30, and C. GAPDH protein, 37 kDa.")
ROR-γt and NKp46 proteins determined by Western blot. GApDH was used to normalize protein concentration as an internal control. Each bar represents the mean value ± standard deviation (SD) of five rats. Results are expressed as area relative units (ARU). A. ROR-γt protein, 63 kDa; ** p ≤ 0.001 control vs. BDL day 8, ** p ≤ 0.001 control vs. BDL day 30. B. NKp46 protein, 47 kDa, ** p ≤ 0.001 control vs. BDL day 30, ** p ≤ 0.001 BDL day 8 vs. BDL day 30, and C. GAPDH protein, 37 kDa.
 and control rats. A. Control groups. B. BDL rats day 8. C. BDL rats day 30. Extrahepatic cholestasis due to prolonged obstruction of bile fow in BDL rats resulted in extensive proliferation of bile ducts in enlarged portal tracts and periductal fibrosis (scale bars: 200 µm).")
Histology findings induced by experimental cholestasis. Representative images of liver sections from bile duct ligation (BDL) and control rats. A. Control groups. B. BDL rats day 8. C. BDL rats day 30. Extrahepatic cholestasis due to prolonged obstruction of bile fow in BDL rats resulted in extensive proliferation of bile ducts in enlarged portal tracts and periductal fibrosis (scale bars: 200 µm).
In this study, the participation of IL-17 and TGF-β1/2 in the pathogenesis of experimental cholestasis was assessed. Various reports dealing with the role of IL-17 in lung, skin, and liver fibrosis have been published.10–12
Heightened gene expression and increase of IL-17 protein level in early and advanced experimental cholestatic fibrosis was observed in our study. These findings have not been previously described in the literature. Accumulation of IL-17 positive cells around the damaged bile ducts in primary biliary cirrhosis (PBC), as well as the ability of biliary epithelial cell cultures stimulated with IL-17 to produce Th17-inducible cytokines (IL-6 and IL-1β) and Th17-maintaining cytokine (IL-23) have been reported by Nakanuma, et al.25 These results suggest that periductal IL-17 secreting cells might facilitate the migration of inflammatory cells around the bile ducts, aggravating the chronic cholangitis in the BDL model. In addition, serum TNF-α, TGF-β1 levels and fibrosis score were significantly decreased in knockout mice with induced cholestatic fibrosis, when compared with the WT mice.18 Moreover, induction of collagen type 1 expression by IL-17 in HSC cultures promoting their activation into fibrogenic myofibroblasts via STAT-3, have been observed in vitro.11
Thus, the above observations indicate that in the BDL model, IL-17 might not only exacerbate the local inflammatory process, but might also contribute to the deposition of extracellular matrix components through collagen production. It is noteworthy to mention that, in our study the increase of IL-17 was more evident at the level of gene expression than at the protein level. This finding might be explained due to the fact that the anti-IL-17 antibody used in our study is designed to recognize total IL-17, and the gene expression was specific for IL-17A.
This cytokine represents the most studied isoform in chronic liver disease. Additionally, a dramatic overexpression of IL-17F mRNAwas also observed in our study (data not shown), evidencing the importance of understanding the biological participation of distinct IL-17 isoforms (A-F) in liver damage.
IL-17 constitutes the major cytokine produced by Th17 cells; however, T cells, CD8+ T cells, γδ T cells, NKT cells, innate lymphoid cells, eosinophils, neutrophils, and monocytes can also produce IL-17. The presence of transcription factors and cytokines, such as STAT3, IL-6, IL-1β, and TGF-β, are required for Th17 differentiation, leading to the induction of the expression of RORγt, a master gene regulator. Likewise, IL-23 expression is necessary for maintaining Th17 phenotype and function.18,19
We analyzed the expression of RORyt as an indicator of the presence of Th17 cells and statistically significant increased expression of RORγt in the liver of cholestatic rats during the chronic stages of liver injury, suggesting that Th17 could be an important source of IL-17 in the BDL model was demonstrated in our study. These results are also supported by the TGF-β1/2 overexpression observed both in the early as well as in the late stages of fibrosis, where TGF-β is essential to Th17 cells differentiation in rodents. A methodological flaw in our study has been the fact that we did not identify the exact cell population producing IL-17; therefore, it will be necessary to plan future studies, which include immunohistochemestry assays in hepatic biopsy or multicolor flow cytometric analysis in isolated hepatic cells, in order to discriminate inflammatory infiltrate from hepatic parenchyma (biliary epithelial cells, Kupffer cells or HSC cells) as a probable source for IL-17.
Parallel to these findings, the decreased expression of NKp46 exhibited in our study suggests that in the BDL model, it might not be a considerable source of IL-17; however, it is likely to be contributing to the exacerbation of the liver damage process.
An interesting antifibrogenic mechanism in the model of CCl4 chronic intoxication, orchestrated by the cytotoxic capacity of NK cells, has been described. In rodents, activation of HSC releases retinoic acid leading to RAE-1 synthesis as a metabolic by-product, sensitizing NK cells through their receptor NKG2D to produce INFγ and TRAIL, inducing the arrest of the HSC cell cycle and death, blocking the perpetuation of hepatic injury.26,27 Future multicolor flow cytometry staining by using a heterogeneus gamma of markers, besides NKp46 (CD16, CD94, CD158, NKG2D, for example), will lead us to identify and quantify intrahepatic NK cells, which will clarify in turn, the participation of NK cells during the whole process of hepatic damage by cholestasis.
On the other hand, it is important to elucidate the cooperative role established between IL-17 and TGF-β1/2 in the development of liver experimental fibrosis in BDL model, since a regulatory role of TGF-β1 in allograft fibrosis has been attributed to IL-17 recently.
TGF-β is a key molecule in diverse fibrotic disorders, it can promote fibroblast proliferation, differentiation, migration, adhesion, and survival. TGF-β can induce cytokine secretion and, most importantly, upregulation of collagen and ECM synthesis, playing a critical role in pathological fibrogenesis.28 The role of TGF-β1 has been well documented in diverse models of acute and chronic hepatic injury. Recently, however, TGF-β2 has been recognized as an important cytokine implicated in the pathophysiology of fibrosis induced by different etiologies. In early and advanced models of renal fibrosis, TGF-β2 expression was markedly increased and specific targeting of TGF-β2 attenuated the renal fibrosis process in experimental diabetes.29,30 Likewise, in ocular fibrosis, TGF-β2 participates in ECM remodeling through the stimulation of the canonical Smad signaling pathway via Smad2/3.9 In this work, we demonstrated that TGF-β1 and TGF-β2 significantly increased during the whole fibrogenic process and, interestingly, TGF-β2 prevailed over TGF-β1 at the messenger and protein levels as well. To our knowledge, these findings have not been reported previously. Chen Taiwan, et al. reported, in three patients with biliary atresia, that TGF-β2 was the most actively transcribed TGF-β gene during the progress of liver fibrosis, while by immunostaining, it was mainly localized in the bile epithelium.7
Kouroumalis, et al. have described intrahepatic bile ductules as a novel source of TGF-β isoforms (1/2/3) in patients with PBC and HCV cirrhosis.31 This finding is relevant due to the displaying features of epitelial-to-mesenchymal transition (EMT) of cholangyiocytes during hepatic fibrosis.31–34
Studies in vitro and in vivo have demonstrated the participation of TGF-β1 in the induction of collagen production as well as in EMT-associated changes in several liver cell types, including cholangiocytes.33,35–37
In the future, the elucidation of TGF-β2 role in EMT will contribute to further understand the hepatic fibrosis process. In the same context, obliteration of TGF-β2 in BDL model would provide useful information regarding the participation of this cytokine in fibrosis progression.
Finally, it is important to point out that inflammation and fibrosis are preponderant mechanisms during the development of the cholestasis. In this context, we might conclude that, based on our present results, TGF-β and IL-17 are important protagonists in the pathophysiology of this disease.
Abbreviations- •
BDL: bile duct ligation.
- •
ECM: extracellular matrix.
- •
HSC: hepatic stellate cells.
- •
IL-17: interleukin 17.
- •
NK: natural killer cells.
- •
PBC: primary biliary cirrhosis.
- •
TGF-β: transforming growth factor beta.



