



Hepatic involvement in AL amyloidosis may present as acute liver failure. Historically, liver transplantation in these cases has achieved poor outcomes due to progress of amyloidosis and non-hepatic organ damage. In the era of bortezomib treatment, the prognosis of AL amyloidosis has been markedly improved and may also result in better post-transplant outcomes. We present a case of isolated acute liver failure caused by AL amyloidosis, bridged to transplantation with bortezomib and treated with sequential orthotopic liver transplantation (OLT) and autologous stem cell transplantation. The patient is in stable remission 3 years after OLT.
Amyloidosis is a systemic disorder characterized by extracellular tissue deposits of fibrils, which are composed of low molecular weight proteins. In AL amyloidosis, the most common form, protein fibrils are derived from the light chains of monoclonal immunoglobulins.1 AL amyloids can accumulate in any organ and, if untreated, may lead to fatal organ failure.2 Hepatic involvement is common but clinically-significant liver disease is usually mild or may go unnoticed.3 In rare cases, however, hepatic amyloidosis may also cause hepatomegaly and hepatocellular decompensation, resulting in liver failure and death.4 The extent of organ involvement affects the outcome and prognosis of AL amyloidosis. In this setting, jaundice carries a very poor prognosis. Few cases of liver transplantation5,6 have been reported, and long-term survival in AL amyloidosis patients has been poor probably because liver transplantation is not curative of the underlying disease and does not address abnormal protein production nor does it prevent extra-hepatic organ damage.5 Here, we report a case of successful liver transplantation followed by high-dose chemotherapy and autologous stem cell transplantation in a patient presenting acute liver failure (ALF) due to AL amyloidosis.
Case ReportA 52-year-old Caucasian male was admitted to the Klinikum Rechts der Isar in Munich in June 2012 with progressive jaundice, clay-colored stool, fatigue, weight loss of about 5 kg, and epigastric pain that persisted for four weeks.
His past medical history included benign prostatic hyperplasia with no previous history of liver disease. General examination showed jaundice and massive hepatomegaly with no other stigmata of chronic liver disease.
Laboratory tests revealed the following: creatinine 80 µmol/L (reference: 62-115 µmol/L), blood urea 5 mmol/L (reference: 2.5-6.5 mmol/L), total bilirubin 140 µmol/L (reference, <21 µmol/L), alkaline phosphatase (aP) 610 U/L (reference, 40-129 U/L), γ-glutamyltransferase (gGT) 979 U/1 (reference, <66 U/L), alanine aminotransferase (ALT) 76 U/L (reference, 10-50 U/L), aspartate aminotransferase (AST) 149 U/1 (reference, 10-50 U/L), serum albumin 43 g/L (reference, 35-50 g/L), INR 1.1. The complete blood count and serum electrolytes were within the normal range. Serologic tests for hepatitis B, hepatitis C and HIV were negative. The quantitative immunoglobulin (Ig) assay showed mild elevation of IgG (1,815 mg/dL) and normal levels of IgM and IgA. Immunofixation identified IgG-gammopathy with kappa light-chain expression. Furthermore, abdominal ultrasonography revealed massive hepato-splenomegaly with no focal liver lesions and no intra- or extrahepatic dilatation of bile-ducts and no ascites.
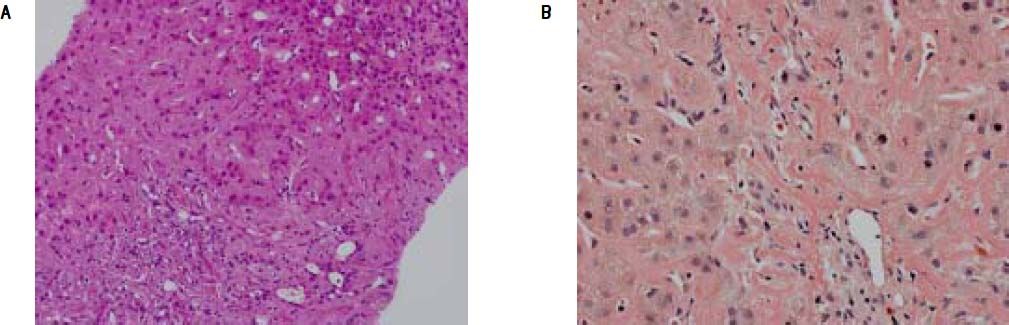
A percutaneous liver biopsy was performed. Light microscopy revealed liver tissue with dilated portal fields with a pronounced ductal reaction, massive canalicular and hepatocellular cholestasis, and amyloid deposition in portal arteries and in the perisinusoidal space of Disse, causing compression of the adjacent hepatocytes Figure (1A). The amyloid deposits were confirmed by Congo red staining (Figure 1B) and were of kappa-light-chain origin as identified by immunohistochemical staining.
 showing perisinusoidal accumulation of amyloid with atrophy of the liver cell plates and narrowing of sinusoids. Pronounced ductular cholestasis is seen at upper right. B. Congo-red stain of the same sample highlighting amyloid deposits in arteries of portal tracts and the perisinusoidal space.")
A. Liver biopsy specimen (H&E) showing perisinusoidal accumulation of amyloid with atrophy of the liver cell plates and narrowing of sinusoids. Pronounced ductular cholestasis is seen at upper right. B. Congo-red stain of the same sample highlighting amyloid deposits in arteries of portal tracts and the perisinusoidal space.
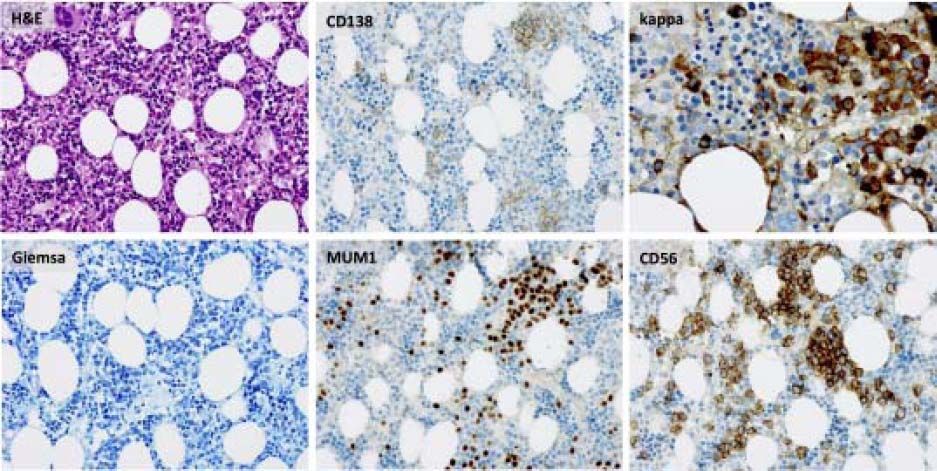
Bone marrow biopsy identified infiltration of atypical plasma cells in the bone marrow with an estimated cellularity of 10% (Figure 2). A bone scan showed no osteolytic lesions, cardiac-MRT showed no signs of a cardiac involvement, and there was no evidence of nephropathy on ultrasound or urinalysis.

Consequently, the patient was diagnosed with systemic AL amyloidosis involving the liver.
In June 2012 induction therapy with Bortezomib (1.3 mg/m2 s.c. on days 1, 4, 8, and 11) and Dexamethasone (40 mg PO on days 1-4 and days 9-12) was started. Despite a relevant decrease in light chain serum levels occurring after the first cycle, deterioration of liver function prevented further rounds of induction therapy, subsequent consolidation therapy with high dose Melphalan, and autologous hematopoietic stem cell transplantation (PB-SCT). The patient developed massive ascites, hepato-renal syndrome and Grade II hepatic encephalopathy. At this time, laboratory results were as follows: creatinine 142 µmol/L (reference: 62-115 µmol/L), total bilirubin 710 µmol/L (reference: <21 µmol/L), aP 473 U/l (reference: 40-129 U/l), gGT 365 U/l (reference: <66 U/l), ALT 34 U/l (reference: 10-50 U/l), AST 86 U/l (reference: 10-50 U/l), serum albumin 31 g/L (reference: 35-50 g/L), INR 1.3.
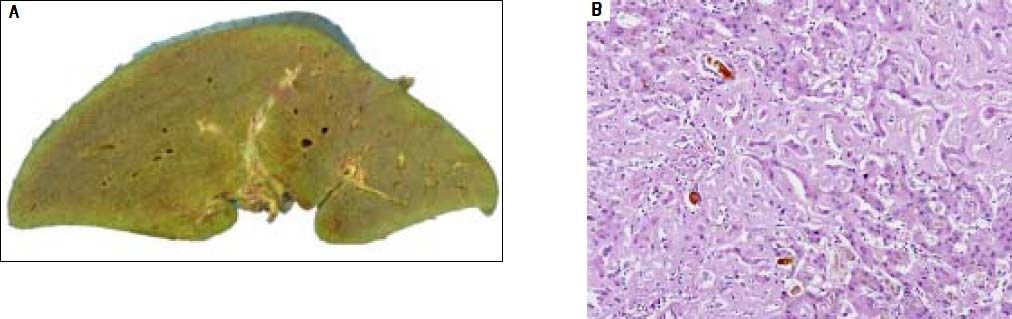
The patient was wait listed for a highly urgent liver transplant, which took place three days later. On pathological examination, the explanted liver showed a massive perisinusoidal and vascular amyloidosis with cholestasis and extensive hepatocyte necrosis (Figures 3A, 3B). The postoperative course was uncomplicated and kidney function improved. An immunosuppressive regimen with mycophenolate mofetil, prednisolone, and tacrolimus was instituted. Four months later, after stabilization of the patient’s condition and normalization of renal function, the patient received two further cycles of Bortezomib and Dexamethason, resulting in a very good partial remission (VGPR). Peripheral blood stem cells were then successfully collected following treatment with cyclophosphamide (2 g/m2) and GCSF, an effective and well-established protocol for hematopoietic stem cell mobilization.7 Thereafter the patient underwent single high-dose melphalan therapy (200 mg/m2) and autologous peripheral blood stem cell transplantation. During treatment with dexamethasone and cyclophosphamide, immunosuppressive treatment was reduced from triple immunosuppression with prednisolone, mycophenolate-mofetil, and tacrolimus to low dose tacrolimus monotherapy aiming at through levels of 2-4 ng/mL. Mycophenolatemofetil was re-instituted 3 weeks after PBSCT and low-dose tacrolimus was continued. The patient tolerated high-dose chemotherapy without any change to the function of the transplanted liver, and complete remission (CR) from AL amyloidosis was achieved. Follow-up percutaneous liver biopsy was done 15 months after OLT and was normal with no evidence of amyloid recurrence or rejection.
 with extensive hepatocyte necrosis (B).")
At the most recent follow up 36 months after OLT, the patient was in a very good general condition and working full-time. Laboratory values were as follows: total bilirubin 14 µmol/L (reference: < 21 µmol/L), aP 81 U/l (reference: 40-129 U/l), gGT 93 U/l (reference: < 66 U/l), ALT 23 U/l (reference: 10-50 U/l), AST 27 U/l (reference: 10-50 U/l), serum albumin 47 g/L (reference: 35-50 g/L), INR 1.1, serum creatinine 106 µmol/L (reference: 62-115 µmol/L). Full blood count and serum electrolytes are normal. The patient continues to be in complete remission from AL amyloidosis and is under dual immunosuppression with mycophenolate mofetil and tacrolimus.
DiscussionHepatic involvement is common in amyloidosis, but it is usually mild and of no clinical significance.8 Only 5% of the patients will present jaundice.9
In a study conducted on 229 patients with primary systemic amyloidosis, Kyle and Greipp reported that 34% of the patients had hepatomegaly; in 16% alkaline phosphatase was elevated and 4% had hyperbilirubinemia. Fatigue and weight loss were the most common symptoms in this study and affected 54 and 42% of patients, respectively.10 The prognosis of patients with cholestatic amyloidosis is usually poor and death occurs a few months after the development of jaundice.11,12 Our patient presented jaundice and deteriorated to acute liver failure, which was also reported to have a poor prognosis and high mortality rate in this setting.5,13,14 AL amyloidosis also occurs in up to 15% of myeloma patients15 and mortality usually is caused by protein deposition in kidney and heart, whereas liver involvement is mostly asymptomatic.3
Median survival of patients with primary hepatic amyloidosis is reported to be 9 months with 5 and 10 years survival of 13 and 1%, respectively. Median survival rates decreased to 1.8 and 3.3 months, respectively, when total bilirubin was more than 1.5 mg/dL(66 µmol/L) or alkaline phosphatase was elevated above 4 times the normal range.3
Treatment outcome in AL amyloidosis depends on the rapid elimination or marked reduction of the amyloid fibril protein with the least possible systemic toxicity.16,17 Melphalan and prednisone were introduced in 1972 as chemotherapy for treatment of amyloidosis.18 Only a minority of patients responded well to this strategy with a median survival of 12-18 months.19 The success of hematopoietic cell transplantation in multiple myeloma led some investigators to apply this therapeutic approach in patients with AL amyloidosis.20 Subsequently high dose intravenous melphalan with autologous stem cell transplantation was introduced as a first line treatment in patients with amyloidosis21 with response rates of 65%.22 Novel agents for treatment of multiple myeloma such as immunomodulatory drugs (IMiDs) like thalidomide and lenalidomide, were introduced. Studies conducted on patients with amyloidosis and thalidomide treatment show hematological response in 48% of the patients but with frequent treatment-related toxicity.23 Lenalidomide in combination with dexamethasone, or with melphalan and dexamethasone achieved two-year event-free survival and survival of 54 and 81%, respectively.24,25 However in patients with amyloidosis, lenalidomide may impair kidney function even in patients with no renal involvement by the amyloidosis with recovery rates reported to be only 44% in treated patients.26 The proteasome inhibitor bortezomib is well-established in the first line treatment of multiple myeloma, both in the transplant and non-transplant setting, and its efficacy in AL amyloidosis has been well documented.27–31 Therefore we started treating our patient with bortezomib, which achieved a rapid and marked decrease of light chain production until the deterioration of the patient’s hepatic function limited further treatment.
There are few reports on patients who underwent liver transplantation due to AL amyloidosis. Patients with this condition deteriorate rapidly and often die before transplantation.8,13,32,33 The recurrence of disease in the graft and the continuous systemic progression also limited the role of organ transplantation in AL amyloidosis.34 Only few cases in the literature achieved long-term survival after liver transplantation. The first case of liver transplantation in a case of hepatic amyloidosis was carried out due to spontaneous splenic rupture, but follow-up liver biopsy showed recurrence of the amyloid in the graft after one year.6
In an analysis of data from the UK National Amyloidosis Centre, Sattianayagam, et al. report on the largest cohort of patients who have undergone liver transplantation for hepatic AL amyloidosis. Nine patients received liver transplants between 1984 and 2009. Of these, long-term survival was reported only for two of three patients receiving stem cell transplantation after liver transplantation. Six patients died within the first year after OLT. Causes of death were intraoperative cardiac decompensation in one patient, sepsis in three patients, and deteriorating renal function and unexplained death in one case. One- and five-year survival was 33% and 22%, respectively.34 A predicted outcome of less than 50% survival at one year after OLT is often regarded as prohibitive, given the scarce supply of donor organs. In our case, with acute hepatic failure progressing despite treatment of the underlying AL amyloidosis, liver transplantation was the only life-saving option for our patient. As there was no evidence of extrahepatic organ involvement, we believed that OLT was likely to provide the prerequisite for subsequent stem cell transplantation. After the advent of proteasome inhibitors, which had not been available at the time of any of the previous published reports, a reasonable prognosis seemed achievable.
In conclusion, despite disappointing results of earlier studies, our case study suggests that OLT may nowadays be a life-saving treatment option in patients with liver failure due to hepatic amyloidosis who have no extra-hepatic organ involvement, and are able to receive high-dose chemotherapy and autologous hematopoetic stem cell transplantation after OLT.
AcknowledgementWe are grateful to Ms Brenda Rascon for help in the preparation of the manuscript.



