




Background and aim. Grazoprevir is an NS3/4A protease inhibitor (PI), while elbasvir is an NS5A inhibitor. We performed this meta-analysis to directly compare grazoprevir plus elbasvir and ribavirin regimen vs. grazoprevir and elbasvir without ribavirin in the treatment of hepatitis C virus genotype 1 infection and to precisely evaluate the efficacy of the latter regimen in cirrhotic, IL28 CC genotype patients and those coinfected with human immunodeficiency virus.
Material and methods. A computer literature search of PubMed, Scopus, EBSCO, Embase, and Cochrane central was conducted. Studies were screened for eligibility. Sustained virologic response (SVR) rates were pooled using OpenMeta [Analyst] software for windows. A subgroup analysis was performed to stratify the treatment efficacy according to the different baseline characteristics of HCV patients.
Results. Eight randomized controlled trials (n = 1,297 patients) were pooled in the final analysis. The overall SVR rate was 96.6% with 95% CI [95.5% to 98%]. For cirrhotic patients, the SVR rate was 95.7% with 95% CI [93.9% to 97.5%] and for non-cirrhotic patients, the SVR rate was 97% with 95% CI [95.9% to 98.4%]. Furthermore, the addition of ribavirin (RBV) to the treatment regimen did not significantly improve the SVR (RR 1.003, 95% CI [0.944 to 1.065]).The dual regimen was effective in patient populations with NS3 resistance-associated substitution (RAS). However, this regimen achieved lower SVR rates (< 90%) in patients with NS5A RAS.
Conclusions. We conclude that the 12-week treatment regimen of the fixed dose combination of grazoprevir plus elbasvir achieved high SVR rates in patients with HCV genotype 1 infection. The addition of ribavirin to this regimen did not add a significant benefit.
Hepatitis C virus (HCV) is a major global health concern that affects more than 71 million individuals (2.8% of the worldwide population).1 Central and East Asia and North Africa/Middle East are estimated to have high prevalence rates of HCV infection (>3.5%), causing approximately 350,000 deaths annually.2,3 Cirrhosis develops in approximately 10% to 15% of individuals with chronic HCV infection and is likely to affect 45% of HCV-infected patients by 2030 without a broader application of antiviral therapy.4 In the USA, 40% of liver transplants are attributed to HCV infection. Moreover, the 5-year cumulative risk of malignant transformation is more than 17% in patients with HCV-associated cirrhosis.5 HCV genotype 1 infection is the most prevalent form of the disease, accounting for 60% of its global burden.6
Before 2013, there was a trend towards using protease inhibitors (PIs) as boceprevir and telaprevir with peg-interferon and ribavirin (RBV) for treating HCV genotype 1 infection.7,8 However, the co-administration of these drugs with peg-interferon and RBV increased the risk of life-threatening adverse events, including seizures, bone marrow suppression and neuropsychiatric problems.9 In the recent few years, the development of direct-acting antiviral (DAA) drugs have revolutionized HCV treatment.
Grazoprevir (MK-5172) is an NS3/4A PI with a potent activity against all HCV genotypes except genotype 3,10 while elbasvir (MK-8742) is an NS5A replication complex inhibitor that has a pangenotypic activity and a high resistance barrier against resistance-associated substitutions (RAS) associated with failure of other NS5A inhibitors as ledipasvir and daclatasvir.11,12 Recently, multiple clinical trials have evaluated the efficacy of a once-daily, fixed-dose combination tablet (grazoprevir plus elbasvir) for the treatment of patients with HCV genotype 1 infection.13–20
A previous meta-analysis (of three clinical trials) by Yao, et al. assessed the efficacy of grazoprevir and elbasvir in HCV-genotype 1 infected patients.11 Some of their findings were later opposed by recent trials,17,18,20 such as the higher response rate in treatment-experienced patients compared to treatment-naïve patients and the inferior SVR rate in patients with IL-28B CC genotype, compared to patients with non-CC genotype. Moreover, they calculated response rates for RBV-treated patients and those without RBV treatment without comparing both arms for significance. Further, they did not assess the efficacy of this regimen in cirrhotic patients and those coinfected with human immunodeficiency virus (HIV).11 We performed this meta-analysis to precisely estimate the SVR achieved by this treatment regimen in HCV-genotype 1 patients with cirrhosis, IL28B genotype variants, prior treatment exposure, and HIV coinfection, as well as to determine the value of adding RBV to this treatment regimen.
Material and MethodsWe followed the PRISMA statement guidelines during the preparation of this review and meta-analysis.21 Moreover, we performed all steps of this review in strict accordance with the Cochrane handbook for systematic reviews of interventions.22 All steps of the study were prespecified, and the protocol was registered on PROSPERO (CRD 42016043723).
Literature search strategyWe conducted a computarized literature search through July 2016, using the following search terms (“grazoprevir” or “MK5172”) and (“elbasvir” or “MK8742”) and (“hepatitis C” OR “HCV”), in six electronic databases, including PubMed, Scopus, EBSCO, Embase, Cochrane Central and Ovid Medline. No language or time restrictions were applied. We also searched clinicaltrials.gov to identify ongoing and unpublished studies. Additionally, we manually scanned the bibliography of included studies.
Eligibility criteriaWe included prospective clinical trials meeting the following inclusion criteria:
- •
Studies involving patients with chronic HCV genotype 1 infection and
- •
Studies assessing the safety or efficacy of grazoprevir plus elbasvir drug regimen, with or without RBV.
We excluded:
- •
Non-randomized studies.
- •
Non-human studies.
- •
Studies from which data cannot be reliably extracted, and
- •
Theses and conference abstracts.
Eligibility screening was performed in two steps, each by two independent reviewers (Menshawy A. and Attia A.):
- •
Screening of titles/abstracts of the retrieved records, and
- •
Full text screening for eligibility to meta-analysis. Disagreements were resolved by discussion with a third reviewer (Abushouk A.I.).
Data extraction
Two independent reviewers (Abushouk A.I. and Ahmed H.) extracted the following data from included studies:
- •
Baseline characteristics of enrolled patients.
- •
General criteria of study design.
- •
Efficacy outcomes, including SVR and virologic relapse, and
- •
Commonly reported adverse events whose incidence was > 5% within the study patients. Disagreements were solved by a third reviewer (Attia A.).
To assess the risk of bias within each included study, two independent authors (Ahmed H. and Attia A.) used the Cochrane risk of bias (ROB) assessment tool, described in chapter 8.5 of the Cochrane handbook for systematic reviews of interventions 5.1.0. The Cochrane ROB assessment tool is designed to detect five types of bias, including selection bias (sequence generation and allocation concealment), performance bias (blinding of participants and investigators), detection bias (blinding of outcome assessors), attrition bias (incomplete outcome data), and reporting bias (selective outcome reporting). Each study is classified in each domain as of low, high, or unclear risk of bias.
Assessing risk of bias across studiesTo assess the risk of bias across included studies, we compared the reported outcomes between all studies to exclude selective reporting of outcomes. According to Egger and colleagues, the assessment of publication bias using the funnel plot method is not reliable when the number of pooled studies is < 10. However, we employed the methods by Harbord, et al. to assess the existence of publication bias.23,24
Measures of treatment efficacy- •
The SVR is defined as undetectable HCV RNA level of < 15 IU/mL, 12 weeks after the end of antiviral therapy.
- •
Virologic relapse is defined as detectable HCV RNA level during follow-up after achieving undetectable levels at the end of treatment.
Sustained virologic response rates were pooled as risk ratios (RR) with 95% confidence intervals in a fixed effect model of meta-analysis. Statistical analysis was performed by OpenMeta[Analyst] software (developed by the Center of Evidence Based Medicine).
Subgroup analysisSubgroup analysis was performed to stratify the treatment efficacy according to:
- •
HCV genotype 1 subtypes.
- •
Presence of cirrhosis.
- •
IL 28B genotype.
- •
Previous treatment with PIs.
- •
Baseline viral load before treatment.
- •
Presence of RASs, and
- •
HIV coinfection.
Heterogeneity was assessed by χ2 test and measured by I2 test. In case of significant heterogeneity (χ2 P < 0.1), the random effects model was employed. Otherwise, the fixed effect model was used. Subgroup and sensitivity analyses were used to resolve the heterogeneity.
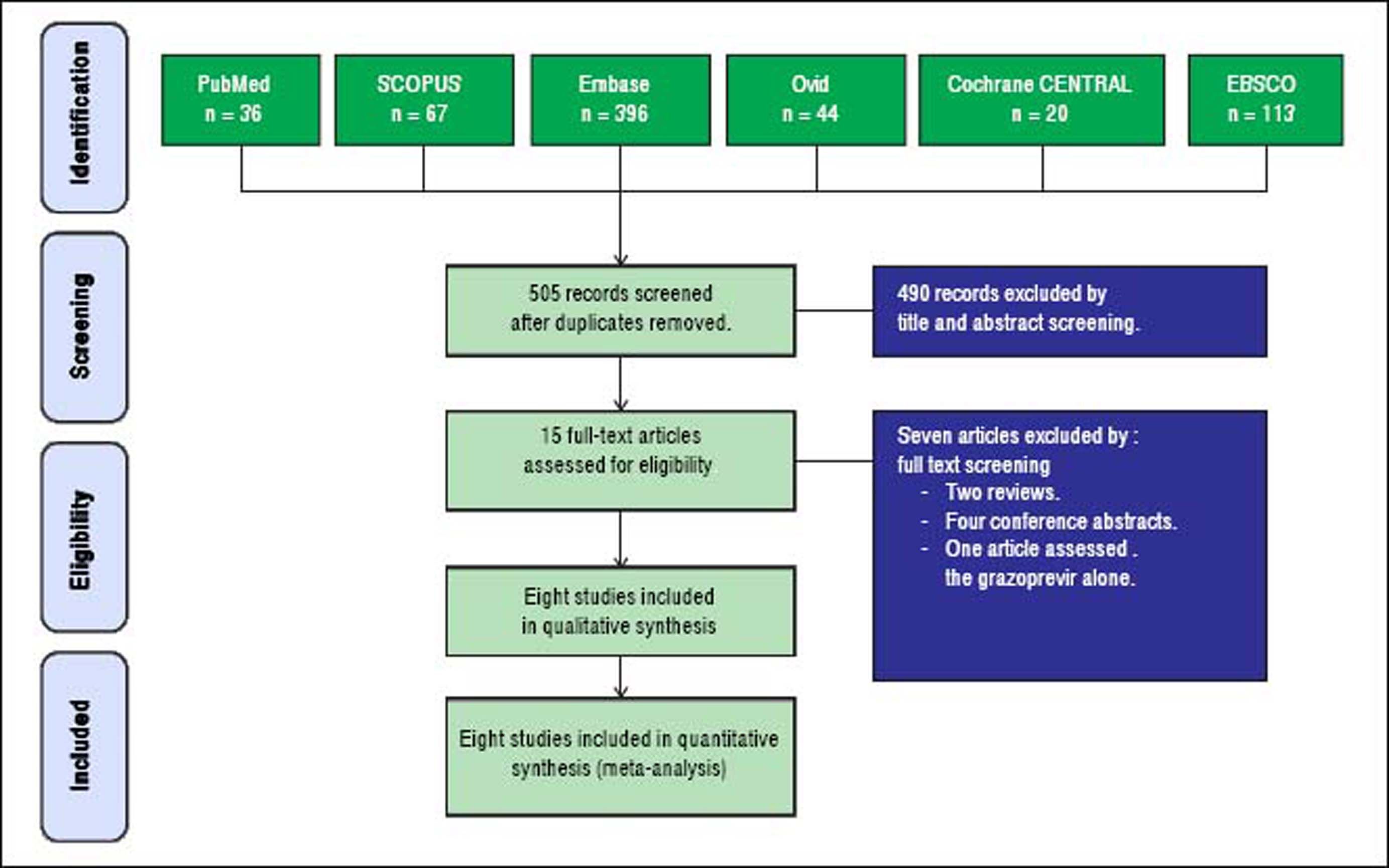
ResultsResults of literature searchOur search retrieved 505 unique records. Fifteen full-text articles were retrieved and screened for eligibility to meta-analysis. Of them, seven articles were excluded and eight RCTs (n = 1,297 patients) were included in the final analysis. The PRISMA flow diagram of study selection is shown in figure 1.
Characteristics of included studies
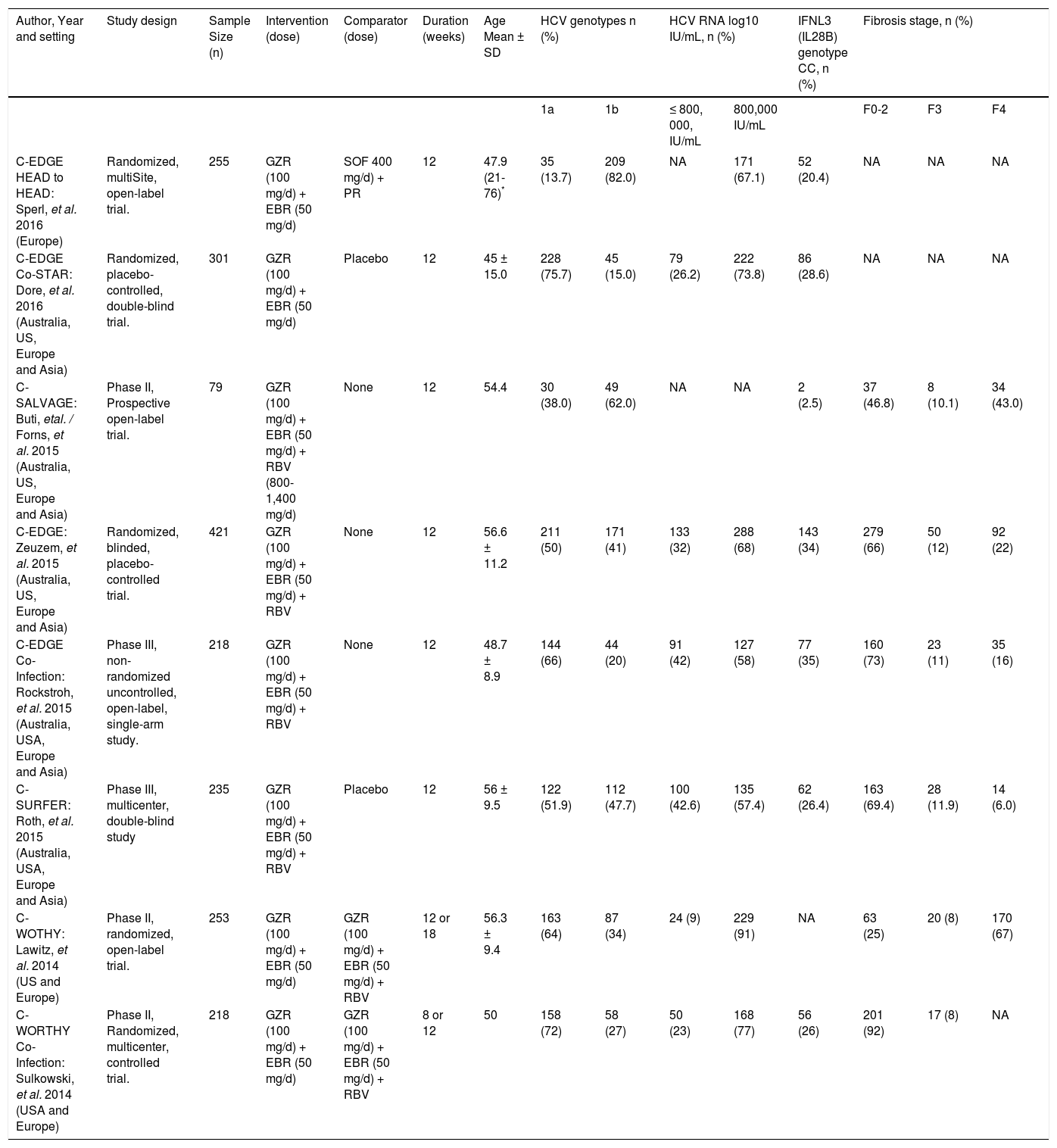
The summary of included studies, their design, baseline characteristics, and key results are shown in table 1. The ROB in the included studies was moderate according to the Cochrane ROB tool. The summary of ROB assessment domains and authors’ judgments with justifications are shown in table 2.
Summary and baseline data of patients in included studies.
| Author, Year and setting | Study design | Sample Size (n) | Intervention (dose) | Comparator (dose) | Duration (weeks) | Age Mean ± SD | HCV genotypes n (%) | HCV RNA log10 IU/mL, n (%) | IFNL3 (IL28B) genotype CC, n (%) | Fibrosis stage, n (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | ≤ 800, 000, IU/mL | 800,000 IU/mL | F0-2 | F3 | F4 | ||||||||
| C-EDGE HEAD to HEAD: Sperl, et al. 2016 (Europe) | Randomized, multiSite, open-label trial. | 255 | GZR (100 mg/d) + EBR (50 mg/d) | SOF 400 mg/d) + PR | 12 | 47.9 (21-76)* | 35 (13.7) | 209 (82.0) | NA | 171 (67.1) | 52 (20.4) | NA | NA | NA |
| C-EDGE Co-STAR: Dore, et al. 2016 (Australia, US, Europe and Asia) | Randomized, placebo-controlled, double-blind trial. | 301 | GZR (100 mg/d) + EBR (50 mg/d) | Placebo | 12 | 45 ± 15.0 | 228 (75.7) | 45 (15.0) | 79 (26.2) | 222 (73.8) | 86 (28.6) | NA | NA | NA |
| C-SALVAGE: Buti, etal. / Forns, et al. 2015 (Australia, US, Europe and Asia) | Phase II, Prospective open-label trial. | 79 | GZR (100 mg/d) + EBR (50 mg/d) + RBV (800-1,400 mg/d) | None | 12 | 54.4 | 30 (38.0) | 49 (62.0) | NA | NA | 2 (2.5) | 37 (46.8) | 8 (10.1) | 34 (43.0) |
| C-EDGE: Zeuzem, et al. 2015 (Australia, US, Europe and Asia) | Randomized, blinded, placebo-controlled trial. | 421 | GZR (100 mg/d) + EBR (50 mg/d) + RBV | None | 12 | 56.6 ± 11.2 | 211 (50) | 171 (41) | 133 (32) | 288 (68) | 143 (34) | 279 (66) | 50 (12) | 92 (22) |
| C-EDGE Co-Infection: Rockstroh, et al. 2015 (Australia, USA, Europe and Asia) | Phase III, non-randomized uncontrolled, open-label, single-arm study. | 218 | GZR (100 mg/d) + EBR (50 mg/d) + RBV | None | 12 | 48.7 ± 8.9 | 144 (66) | 44 (20) | 91 (42) | 127 (58) | 77 (35) | 160 (73) | 23 (11) | 35 (16) |
| C-SURFER: Roth, et al. 2015 (Australia, USA, Europe and Asia) | Phase III, multicenter, double-blind study | 235 | GZR (100 mg/d) + EBR (50 mg/d) + RBV | Placebo | 12 | 56 ± 9.5 | 122 (51.9) | 112 (47.7) | 100 (42.6) | 135 (57.4) | 62 (26.4) | 163 (69.4) | 28 (11.9) | 14 (6.0) |
| C-WOTHY: Lawitz, et al. 2014 (US and Europe) | Phase II, randomized, open-label trial. | 253 | GZR (100 mg/d) + EBR (50 mg/d) | GZR (100 mg/d) + EBR (50 mg/d) + RBV | 12 or 18 | 56.3 ± 9.4 | 163 (64) | 87 (34) | 24 (9) | 229 (91) | NA | 63 (25) | 20 (8) | 170 (67) |
| C-WORTHY Co-Infection: Sulkowski, et al. 2014 (USA and Europe) | Phase II, Randomized, multicenter, controlled trial. | 218 | GZR (100 mg/d) + EBR (50 mg/d) | GZR (100 mg/d) + EBR (50 mg/d) + RBV | 8 or 12 | 50 | 158 (72) | 58 (27) | 50 (23) | 168 (77) | 56 (26) | 201 (92) | 17 (8) | NA |
Results of risk of bias assessment.
| C-EDGE Co-STAR19 | Riks of Bias | Author judgment |
|---|---|---|
| Random sequence generation (selection bias) | Unclear | Not described. |
| Allocation concealment (selection bias) | Low risk | Randomization was done centrally through an interactive voice and integrated Web response system. |
| Blinding of participants and personnel (performance bias) | High risk | Double-blinded for 12 week, followed by 4 week of follow up, then 12 week of open-label treatment. |
| Blinding of outcome assessment (detection bias) | High risk | Not described. |
| Incomplete outcome data (attrition bias) | Low risk | Thirty two patients were lost during follow up period, 10%. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study was funded by Merck & Co., Inc., Kenilworth, New Jersey, USA. |
| C-EDGE HEAD to HEAD20 | Risk of Bias | Author judgment |
| Random sequence generation (selection bias) | Unclear | Not described. |
| Allocation concealment (selection bias) | low risk | Eligible patients were randomized 1:1 by using an interactive voice response. |
| Blinding of participants and personnel (performance bias) | High risk | “We conducted a randomized, multisite, open-label trial”. |
| Blinding of outcome assessment (detection bias) | Unclear | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Two patients were lost during the follow-up period, < 10%. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. |
| C-SALVAGE51 | Risk of Bias | Author judgment |
| Random sequence generation (selection bias) | Unclear | Not described. |
| Allocation concealment (selection bias) | Unclear | Not described. |
| Blinding of participants and personnel (performance bias) | High risk | Open label study. |
| Blinding of outcome assessment (detection bias) | Unclear | Not described. |
| Incomplete outcome data (attrition bias) | Low risk | One patient was lost during the follow-up period, 1%. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study was sponsored and funded by Merck & Co, Inc. |
| C-EDGE Co-Infection17 | Risk of Bias | Quotations |
| Random sequence generation (selection bias) | Unclear | Not described. |
| Allocation concealment (selection bias) | Unclear | Not described. |
| Blinding of participants and personnel (performance bias) | High risk | Open label study. |
| Blinding of outcome assessment (detection bias) | Unclear | Not described. |
| Incomplete outcome data (attrition bias) | Low risk | All patients completed the follow-up period. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study was funded by Merck Sharp & Dohme Corp. |
| C-SURFER18 | Risk of Bias | Quotations |
| Random sequence generation (selection bias) | Low risk | Randomization was done centrally with an interactive voice response system. |
| Allocation concealment (selection bias) | Low risk | All clinical supplies were provided by Merck & Co., Inc. Patients, investigators, and site personnel were masked to the treatment assignment. |
| Blinding of participants and personnel (performance bias) | Low risk | Double-blinded study. |
| Blinding of outcome assessment (detection bias) | Unclear | Not described. |
| Incomplete outcome data (attrition bias) | Low risk | Thirteen patients were lost during the follow-up period, 5%. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study was funded by Merck Sharp & Dohme Corp. |
| C-EDGE16 | Risk of Bias | Quotations |
| Random sequence generation (selection bias) | Low risk | Patients were randomly assigned in a 3:1 ratio to receive immediate or deferred therapy with grazoprevir– elbasvir through a central interactive voice response system according to a computer-generated random allocation schedule. |
| Allocation concealment (selection bias) | Low risk | Patients were randomly assigned in a 3:1 ratio. |
| Blinding of participants and personnel (performance bias) | Low risk | International, randomized, blinded, placebo-controlled. |
| Blinding of outcome assessment (detection bias) | Low risk | Patients, clinical site, and sponsor personnel were blinded to treatment assignment. |
| Incomplete outcome data (attrition bias) | Low risk | Ten patients were lost during the follow-up period, 2%. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study was sponsored and funded by Merck & Co. |
| C-WORTHY14 | Risk of Bias | Quotations |
| Random sequence generation (selection bias) | Unclear | Not described. |
| Allocation concealment (selection bias) | High risk | The investigators and patients were masked to the duration of therapy from randomization to week 12, but were not masked to ribavirin allocation; the study funder (including statisticians) was not masked to treatment allocation or duration. |
| Blinding of participants and personnel (performance bias) | High risk | Open label study. |
| Blinding of outcome assessment (detection bias) | Unclear | Not described. |
| Incomplete outcome data (attrition bias) | Low risk | Fourteen patients were lost during follow up period, 5%. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study funded by Merck & Co, Inc. |
| C-WORTHY Co-Infection15 | Riks of Bias | Quotations |
| Random sequence generation (selection bias) | Unclear | Not described. |
| Allocation concealment (selection bias) | Low risk | Randomization occurred centrally with an interactive Voice response system. |
| Blinding of participants and personnel (performance bias) | High risk | Open label study |
| Blinding of outcome assessment (detection bias) | Low risk | The patients, investigators, and study site personnel were masked to the treatment group assignments. |
| Incomplete outcome data (attrition bias) | Low risk | Twelve patients were lost during the follow-up period, 5%. |
| Selective reporting (reporting bias) | Low risk | All outcomes of interest were reported. |
| Other bias | High risk | This study was funded by AbbVie, Boehringer Ingelheim, Bristol- Meyers Squibb, and Gilead; and personal fees from AbbVie, Bristol-Meyers Squibb, Gilead, Janssen, and Tobira outside the submitted work. |
- •
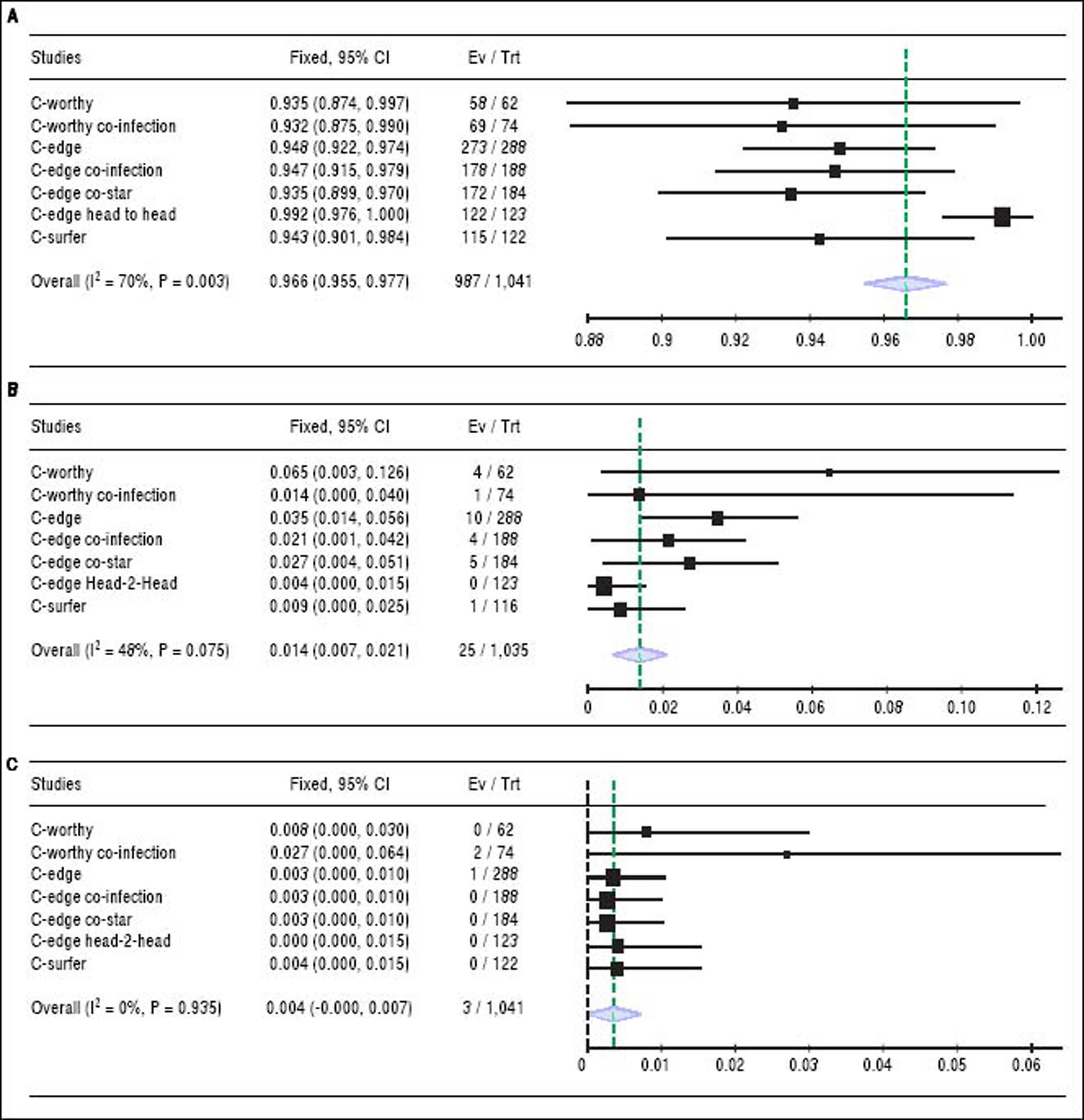
Sustained virologic response. The pooled SVR rate for grazoprevir plus elbasvir was 96.6% with (95% CI [95.5% to 98%], P < 0.001, Figure 2A); pooled studies were heterogenous (P < 0.01; = 70%). The present heterogeneity was best resolved by exclusion of the C-EDGE HEAD to HEAD study, with a SVR rate of 94.3% and 95% CI [92.8% to 95.8%]; pooled studies were homogenous (P = 0.98; = 0%). There was no evidence of publication bias according to Harbord, et al. (P = 0.13).
 Figure 2.
Figure 2.A. Shows forest plots of pooled SRV rates of 12-week regimen of grazoprevir plus elbasvir. B. Forest plots of pooled virologic relapse rates of 12-week regimen of grazoprevir plus elbasvir. C. Forest plots of pooled virologic breakthrough rates of 12-week regimen of grazoprevir plus elbasvir with 95% CI. SVR: Sustained virologic response. CI: Confidence interval.
(0.35MB). - •
Virologic relapse. The relapse rate for grazoprevir plus elbasvir was 1.4% with (95% CI [0.7% to 2.1%], P < 0.001, Figure 2B); pooled studies were heterogenous (P= 0.075; I2 = 48%). Heterogeneity was best resolved by excluding the same study (C-EDGE HEAD to HEAD). Following correction of heterogeneity, the relapse rate was 2.1% with 95% CI of [1.2% to 3%]; pooled studies were homogenous (P = 0.28; I2 = 20%).
- •
Virologic breakthrough. The virologic breakthrough for grazoprevir plus elbasvir was 0.4% with (95% CI [0% to 0.7%], P = 0.53, Figure 2C); pooled studies were homogenous (P = 0.93; I2 = 0%).
- •
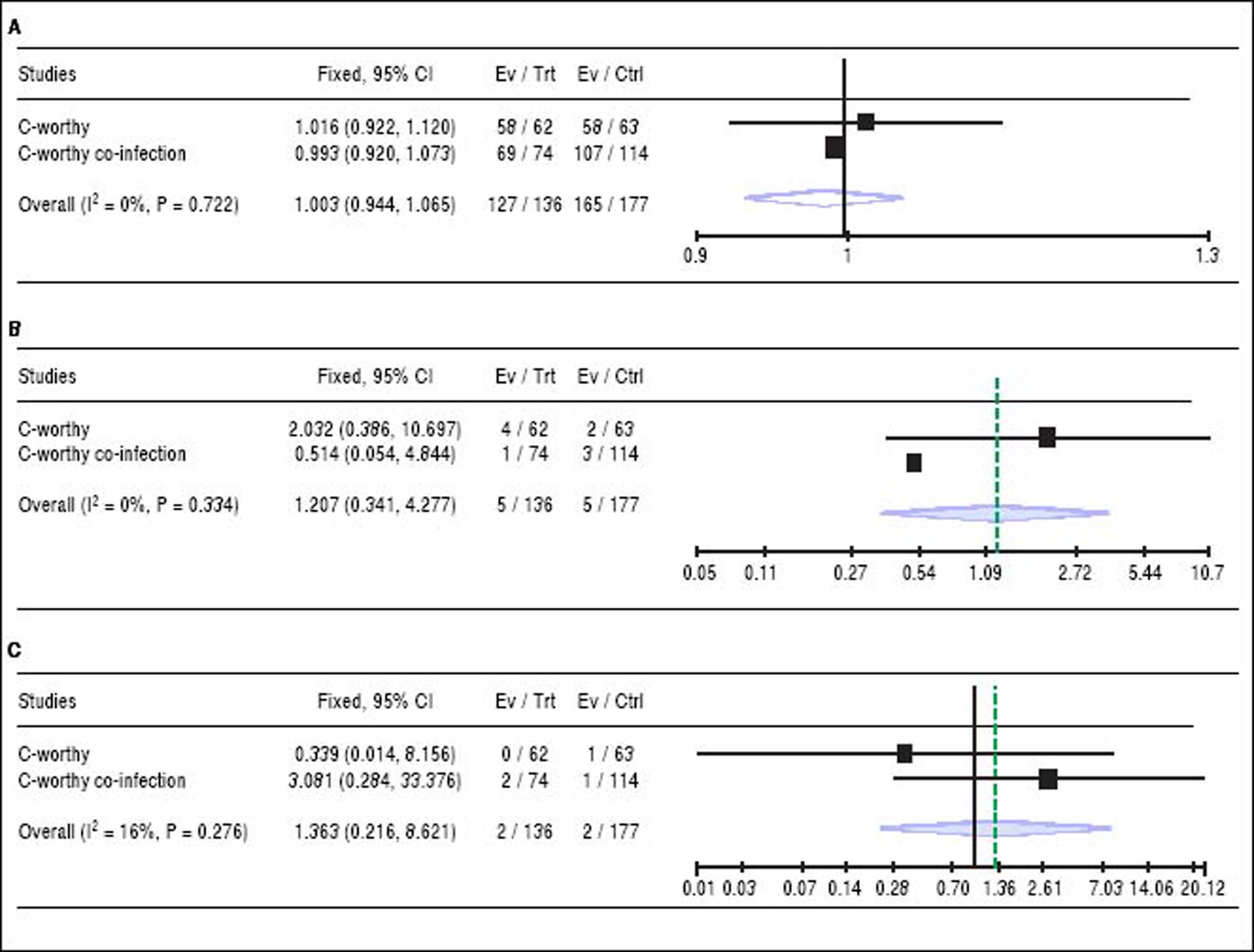
The addition of Ribavirin to the treatment regimen. Two studies14,15 reported the efficacy of the treatment regimen with and without RBV. The pooled RR showed that adding RBV to the grazoprevir plus elbasvir did not increase the SVR (RR 1, 95% CI [0.94 to 1.06], P > 0.05, Figure 3A), or reduce virologic relapse (RR 1.20, 95% CI [0.34 to 4.27], P > 0.05, Figure 3B), and virologic breakthrough rates (RR 1.36, 95% CI [0.21 to 8.6], P > 0.05, Figure 3C). For all comparisons, pooled studies were homogenous (χ2 P < 0.1).
 Figure 3.
Figure 3.A. Forest plots of pooled risk ratio of SRV rates of 12-week regimen of grazoprevir plus elbasvir with or without RBV. B. Forest plots of risk ratio of the virologic relapse rates of 12-week regimen of grazoprevir plus elbasvir with or without RBV. C. Forest plots of pooled risk ratio of the virologic breakthrough rates of 12-week regimen of grazoprevir plus elbasvir with or without RBVwith 95% CI. SVR: Sustained virologic response. CI: Confidence interval.
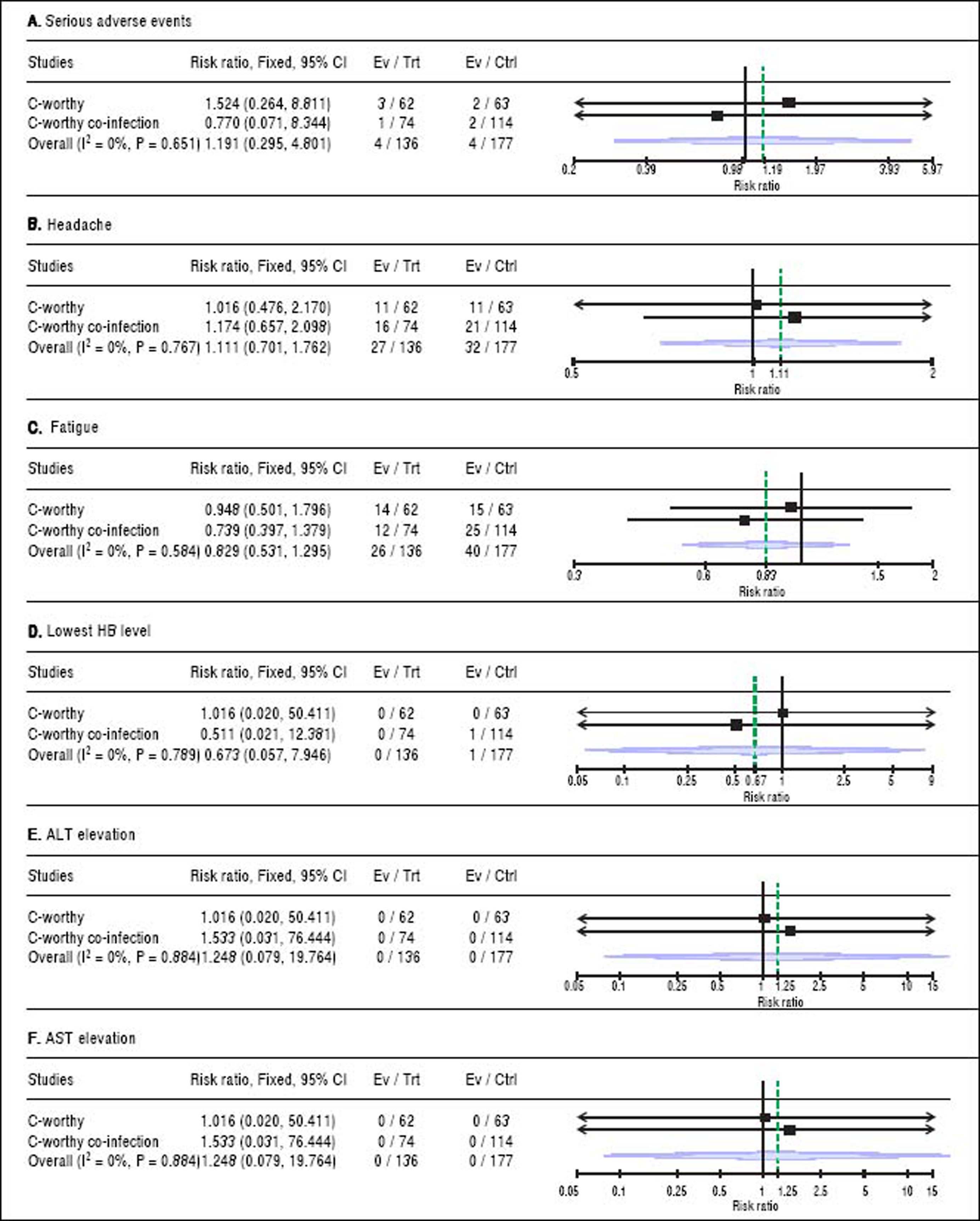
(0.23MB).For safety outcomes, the pooled RR showed no significant difference between grazoprevir plus elbasvir group and grazoprevir plus elbasvir and RBV group in term of serious adverse events (RR = 1.19, 95% CI = [0.29 to 4.80], p = 0.65), headache (RR = 1.11, 95% CI = [0.70 to 1.76], p = 0.76), fatigue (RR = 0.82, 95% CI = [0.53 to 1.29], p = 0.58), lowest Hb level (< 8.5 g/dL) on treatment (RR = 0.67, 95% CI = [0.05 to 7.94], p = 0.789), ALT elevation (> 2.5x baseline) on treatment (RR = 1.24, 95% CI = [0.07 to 9.76], p = 0.88), and AST elevation (> 2.5 x baseline) on treatment (RR = 1.24, 95% CI = [0.07 to 19.76], p = 0.88). For all safety comparisons, pooled studies were homogeneous (χ2 P > 0.1). See figure 4.
- •
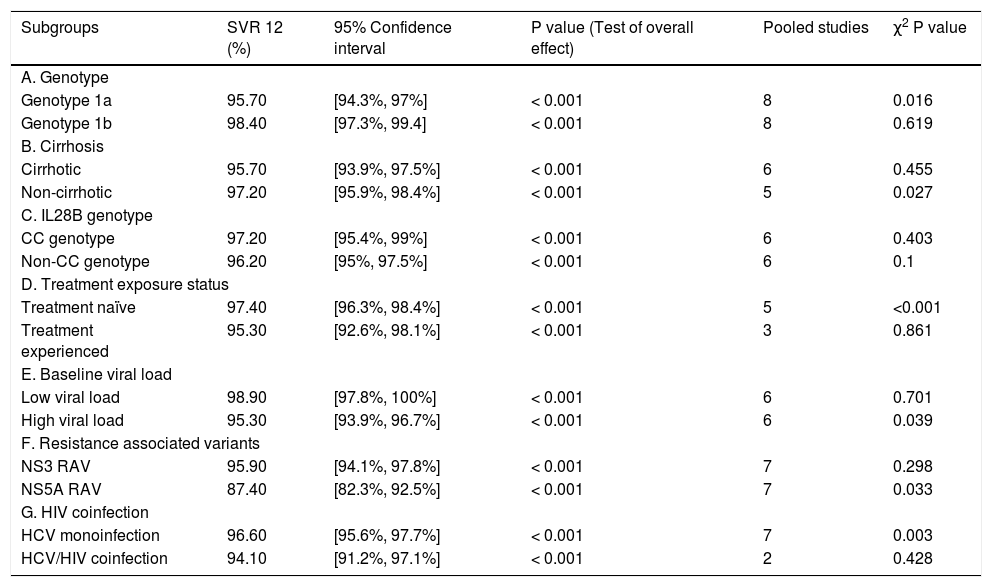
Stratification analysis (genotype 1 subtypes: 1a and 1b). All the eight RCTs13–20 reported on the efficacy of the treatment regimen for genotype 1 subgroups (1a and 1b). The stratification analysis showed that the treatment regimen achieved a SVR rate of 95.7% with 95% CI [94.3% to 97%] in patients with genotype 1a and a SVR rate of 98.4% with 95% CI [97.3% to 99.4%] in patients with genotype 1b.
- •
Stratification analysis (cirrhotic and non-cirrhotic patients). Of the eight analyzed RCTs, six RCTs reported on the efficacy of the regimen in the cirrhotic patients13–18 and five RCTs reported on its efficacy in non-cirrhotic patients.13,14,16–18 The stratification analysis showed that the fixed dose combination of grazoprevir plus elbasvir achieved a SVR rate of 95.7% with 95% CI [93.9% to 97.5%] in cirrhotic patients and a SVR rate of 97.2% with 95% CI [95.9% to 98.4%] in non-cirrhotic patients.
- •
Stratification analysis (IL28B genotypes: CC and non-CC). Of the eight analyzed RCTs, six RCTs reported on the efficacy of the regimen according to IL28B genotype.13–18 The stratification analysis showed that the fixed dose combination of grazoprevir plus elbasvir achieved a SVR rate of 97.2% with 95% CI [95.4% to 99%] in IL28B CC genotype and a SVR rate of 96.2% with 95% CI [95% to 97.5%] in IL28B non-CC genotype.
- •
Stratification analysis (treatment naïve and treatment experienced). Of the eight analyzed RCTs, five RCTs reported on the efficacy of the regimen in treatment naïve patients14–18 and three RCTs reported on its efficacy in treatment experienced patients.13,14,18 The stratification analysis showed that the fixed dose combination of grazoprevir plus elbasvir achieved a SVR rate of 97.4% with 95% CI [96.3% to 98.4%] in treatment naïve patients and a SVR rate of 95.3% with 95% CI [92.6% to 98.1%] in treatment experienced patients.
- •
Stratification analysis according to baseline viral load. Of the eight analyzed RCTs, six RCTs13–18 reported on the efficacy of the regimen in patients with low baseline viral load (≤ 800,000 IU/mL) and high baseline viral load (> 800,000 IU/mL). The stratification analysis showed that the fixed dose combination of grazoprevir plus elbasvir achieved a SVR rate of 98.9% with 95% CI [97.8% to 100%] in patients with low baseline viral load and a SVR of 95.3% with 95% CI [93.9% to 96.7%] in those with high baseline viral load.
- •
Stratification analysis (NS3/NS5A Resistance associated substitution). Of the eight analyzed RCTs, seven RCTs13–18,20 reported on the efficacy of the treatment regimen in patients with NS3 or NS5A RASs. The stratification analysis showed that the fixed dose combination of grazoprevir plus elbasvir achieved a SVR rate of 95.9% with 95%CI [94.1% to 97.8%] in patients with NS3 RAS and a SVR rate of 87.4% with 95% CI [82.3% to 92.5%] in those with NS5A RAS.
- •
Stratification analysis (HCV monoinfection and-HCV/HIV coinfection). Of the eight analyzed RCTs, seven RCTs reported on the efficacy of the treatment regimen in patients with HCV monoinfection13–16,18–20 and two RCTs reported the efficacy in patients with HCV/HIV coinfection.15,17 The stratification analysis showed that the fixed dose combination of grazoprevir plus elbasvir achieved a SVR rate of 96.6% with 95% CI [95.6% to 97.7%] in patients with HCV monoinfection and a SVR of 94.1% with 95% CI [91.2% to 97.1%] in those with HCV/HIV coinfection. A summary of stratification analysis is shown in table 3.
Table 3..SVR12 rates in patient subgroups.
Subgroups SVR 12 (%) 95% Confidence interval P value (Test of overall effect) Pooled studies χ2 P value A. Genotype Genotype 1a 95.70 [94.3%, 97%] < 0.001 8 0.016 Genotype 1b 98.40 [97.3%, 99.4] < 0.001 8 0.619 B. Cirrhosis Cirrhotic 95.70 [93.9%, 97.5%] < 0.001 6 0.455 Non-cirrhotic 97.20 [95.9%, 98.4%] < 0.001 5 0.027 C. IL28B genotype CC genotype 97.20 [95.4%, 99%] < 0.001 6 0.403 Non-CC genotype 96.20 [95%, 97.5%] < 0.001 6 0.1 D. Treatment exposure status Treatment naïve 97.40 [96.3%, 98.4%] < 0.001 5 <0.001 Treatment experienced 95.30 [92.6%, 98.1%] < 0.001 3 0.861 E. Baseline viral load Low viral load 98.90 [97.8%, 100%] < 0.001 6 0.701 High viral load 95.30 [93.9%, 96.7%] < 0.001 6 0.039 F. Resistance associated variants NS3 RAV 95.90 [94.1%, 97.8%] < 0.001 7 0.298 NS5A RAV 87.40 [82.3%, 92.5%] < 0.001 7 0.033 G. HIV coinfection HCV monoinfection 96.60 [95.6%, 97.7%] < 0.001 7 0.003 HCV/HIV coinfection 94.10 [91.2%, 97.1%] < 0.001 2 0.428



This study expands the literature by providing class one evidence about the safety and efficacy of grazoprevir plus elbasvir treatment regimen for patients with HCV genotype 1 infection. Moreover, it presents some interesting findings:
- •
The addition of RBV did not significantly increase the efficacy of the grazoprevir plus elbasvir combination.
- •
This regimen was effective for both cirrhotic and non-cirrhotic patients, and
- •
Treatment-experienced patients or those with NS3 RAS or HCV/HIV coinfection can be treated successfully with the 12-week combination of grazoprevir plus elbasvir.
Our analysis showed a significant heterogeneity in the pooled SVR and relapse rates. This heterogeneity in both outcomes was best resolved by sensitivity analysis, excluding the C-EDGE HEAD to HEAD study. This could be explained by the patients of the C-EDGE HEAD to HEAD study having favorable baseline profiles (lower baseline viral load, less percentage of cirrhotic patients, and less RAS) than those in other studies.
The addition of ribavirin to the treatment regimenRibavirin is often used as an adjuvant drug in several HCV treatment regimens because it can reduce the risk of viral relapse.25–27 However, the published literature highlights its association with several adverse events, particularly hemolytic anemia, abdominal discomfort, rash and pruritus.28–30 Moreover, the value of adding RBV to the newly DAA regimens is not established. Two RCTs (C-WORTHY and C-WORTHY Co-INFECTION) compared the efficacy of grazoprevir plus elbasvir with vs. without RBV. Both studies showed that the addition of RBV yields no significant efficacy benefits.14,15
A previous study by our team showed that the addition of RBV does not increase the efficacy of ledipasvir plus sofosbuvir in patients with HCV genotype 1 infection.31 On the contrary, recent studies on other treatment regimens advocate that RBV might have a therapeutic role, especially in the presence of RASs.32,33 This notion should be investigated for the treatment regimen of grazoprevir plus elbasvir. In addition, other variables as fibrosis stage, HCV genotype, and presence of RAS should be incorporated in the decision of adding RBV to the treatment regimen.34
The efficacy of the treatment regimen in HCV genotype 1 subtypesInterestingly, our meta-analysis showed that patients with HCV genotype 1a infection achieved relatively lower SVR rates (95.7%) than those with genotype 1b (98.4%). The literature suggests that the presence of RASs, which were common in the analyzed RCTs (580/1,113), might lower the efficacy of the treatment regimen in patients with genotype 1a than those with genotype 1b.35,36 This correlation has not been established for this drug regimen; therefore, future studies are encouraged to investigate this hypothesis.
The efficacy of the treatment regimen in cirrhotic patientsIt is known that the efficacy of the treatment regimen might decrease substantially in cirrhotic patients; this was well-established with many treatment regimens as simeprevir plus peginterferon and RBV37,38 and ABT-450/ r-Ombitasvir-dasabuvir with RBV.39 Our analysis shows that grazoprevir plus elbasvir can achieve high SVR rates in both cirrhotic (95.7%) and non-cirrhotic patients (97.2%).
The efficacy of the treatment in presence of resistance associated variantsNS3 RASs can negatively influence the efficacy of other protease inhibitors-based regimens as simeprevir plus peginterferon and RBV (SVR of 84% without NS3 RASs vs. 58% with NS3 RASs).40 Our results showed that the presence of NS3 RAS does not influence SVR rates for grazoprevir plus elbasvir drug regimen. This supports the results of in-vitro studies where NS3 mutations did not decrease the efficacy of grazoprevir.10
On the other hand, our analysis showed that patients who had NS5A RAS at baseline achieved low SVR rate (83.2%). The most commonly reported NS5A RAS in the included studies was NS5A-Y93H. A similar effect was noted for the NS5A RASs in the ION-3 study where naïve patients received sofosbuvir plus ledipasvir.41 However, extension of therapy to 16 or 18 weeks with the addition of weight-based RBV is recommended by Thompson, et al. to improve SVR rates among prior non-responders with NS5A RASs.42
The efficacy of the treatment regimen in HIV-coinfected patientsOur analysis showed that grazoprevir plus elbasvir regimen is effective in both HCV monoinfected and HCV/ HIV co-infected patients (SVRof 96.6 and 94.1%, respectively). This supports the growing body of evidence that both patient populations achieve high responses to interferon-free oral DAA regimens.43–46 It should be mentioned that the C-WORTHY Co-Infection study found a trend towards a higher SVR rate in the monoinfected patients than HIV-coinfected patients when baseline viral load exceeded 107 IU/mL.15 This observation goes with the results of former studies where the effect of HIV coinfection became prominent when the baseline viral load exceeded two million copies/mL.47,48 However, recent guidelines make no distinction between HCV monoinfected and HCV/HIV coinfected patients.49
The treatment duration with grazoprevir plus elbasvir combinationCurrent data support the use of a twelve weeks’ regimen of this combination. The C-WORTHY trial demonstrated that treatment with grazoprevir plus elbasvir for 12 or 18 weeks achieved similarly high SVR rates.14 In the C-WORTHY Co-Infection study, using the same regimen for eight weeks achieved lower SVR rates than the 12 weeks’ regimen (80% and 95% respectively). It also caused an unacceptably high relapse rates (4 to 12% for the eight weeks’ arm vs. 1% for the 12 weeks’ arm).15 Achieving shorter treatment durations by using additional DAA drugs (as sofosbuvir) is currently under investigation (C-SWIFT study).50 Data for SVR at 24 weeks post-treatment were provided in the C-SALVAGE Estudy51 for the dual regimen without RBV (96.2%, equal to SVR12 of 96.2%) and the C-EDGE CO-STAR study19 for the triple regimen with RBV (85.9%, in comparison to SVR12 of 93.5%).
Completeness of evidenceWe performed a comprehensive literature search of major electronic databases to include all relevant reports. The percentage of incomplete data in the analyzed reports was < 5%; of the eight included RCTs, there were 34 (2.6%) withdrawals after randomization, of which only six withdrawals were described as discontinuations due to adverse events. Moreover, the authors are aware of other ongoing phase III trials, NCT02252016, NCT02105701, NCT02332707, and NCT02251990 evaluating the efficacy of grazoprevir plus elbasvir regimen in chronic HCV patients.
Limitations of the studyOur meta-analysis has some limitations:
- •
Five of the eight included RCTs had an open-label design, opening a possible source of performance bias in these studies,
- •
Most of the cirrhotic patients, reported in these RCTs, were compensated. Therefore, our results cannot be generalized to decompensated patients,
- •
The included RCTs included patients from Europe, Australia, and USA with lack of representation from Asia, Africa, and Latin America,
- •
Only two studies assessed the effect of HIV coinfection15,17 or adding RBV14,15 on the efficacy of the grazoprevir plus elbasvir regimen, and finally
- •
Our meta-analysis analyzed the published data collectively; we did not have access to individual patient data; therefore, we could not investigate whether the addition of RBV was beneficial for patients with RAS and whether HCV/HIV coinfected patients with a high baseline viral load achieve lower SVR rates with the present regimen.
Future studies overcoming these limitations are recommended.
ConclusionThe 12-week treatment regimen of the fixed dose combination of grazoprevir plus elbasvir achieved high SVR rates in patients with HCV genotype 1 infection. The addition of RBV to this regimen was not beneficial. The regimen was effective in patient populations with/without cirrhosis, with CC/non-CC IL28B genotypes, with/without prior treatment exposure, and in those with NS3 RAS. The only stratum where this regimen achieved lower SVR (< 90%) was that of patients with NS5A RAS. Therefore, newer regimens with a higher resistance barrier should be developed.
Key Points- •
Fixed dose combination of grazoprevir plus elbasvir achieved high SVR rates in patients with HCV genotype 1 infection.
- •
The addition of RBV to grazoprevir plus elbasvir was not beneficial in term of SVR and relapse rates.
- •
This regimen was effective in patient populations with/without cirrhosis, with CC/non-CC IL28B genotypes, with/without prior treatment exposure, and in those with NS3 RAV.
- •
The only stratum where this regimen achieved lower SVR (<90%) was that of patients with NS5A RAS.
- •
PI: Protease inhibitor.
- •
RAS: Resistance associated substitution.
- •
RBV: Ribavirin.
- •
SVR: Sustained viral response.
None to declare.
Funding SourcesNone to declare.
PROSPERO identifier: CRD 42016043723.
AcknowledgementsNone to declare.



