



Liver and eyes are interlinked to each other in various medical conditions. There are certain ocular findings which directly indicate specific liver disorders. Thus, it becomes critical to identify disorders of liver and eyes early in the course of illness, so that prompt management may be initiated before the commencement of complications. It is highly advantageous in metabolic liver disorders as it offers prognostic value and spares the patient of unnecessary invasive and detailed work up. However, due to its silent and heterogeneous presentation, it is often unrecognized and ignored. Eye abnormalities could be due to, either direct toxic effects of abnormal metabolites, excess of normal metabolites, or by deficient energy metabolism. A number of inherited liver conditions have associated ocular lesions such as Kayser–Fleischer rings in Wilson's disease, posterior embryotoxon or optic drusen in Alagille's syndrome, and cherry-red spot in Niemann–Pick's type A. A thorough eye examination is important in distinguishing between several different forms of familial intrahepatic cholestasis which are associated with anomalies of the heart, bones, or kidneys. Early diagnosis is important, as in most cases, dietary restriction and early therapy prevents the onset of disability. The aim of this review is to sensitize and make pediatricians, hepatologists and ophthalmologists aware of specific ocular findings, suggestive of certain hepatobiliary disorders, thus helping in early referral. The pediatric and adult literature was thoroughly reviewed to organize the present review.
The liver is the center for all metabolic pathways in the body. It plays a pivotal role in metabolizing all macro and micronutrients in the body. There are several known liver metabolic defects which can lead to either abnormal or excess/deficient metabolites [1]. Most are due to a defect in an enzyme or transport protein, resulting in blocking a certain metabolic pathway. The effects are due to toxic accumulations of substrates before the block, intermediates from alternative metabolic pathways, and/or defects in energy production and utilization caused by deficiency of products beyond the block. These abnormal metabolites can either deposit directly or cause an indirect effect on several organ systems, eyes being an important one [1–3]. The age of onset of ocular abnormalities in metabolic liver disease is variable, but often begins in childhood, infancy, or may present at birth. Eye examination is important in evaluating and discriminating among several forms of familial intrahepatic cholestasis and other chronic liver disorders. Hence ocular examination becomes mandatory in all suspected cases of liver metabolic disorders where ocular lesions co-exist. Early diagnosis is important, as in most cases, dietary restriction and early therapy prevents the onset of complications. Moreover, ophthalmic monitoring in certain liver diseases like autoimmune hepatitis is crucial to recognize the side effects of steroids during management such as ocular hypertension, glaucoma and cataract. Hereby, in this review we will discuss the important ocular signs, which are clinically relevant in the setting of liver diseases.
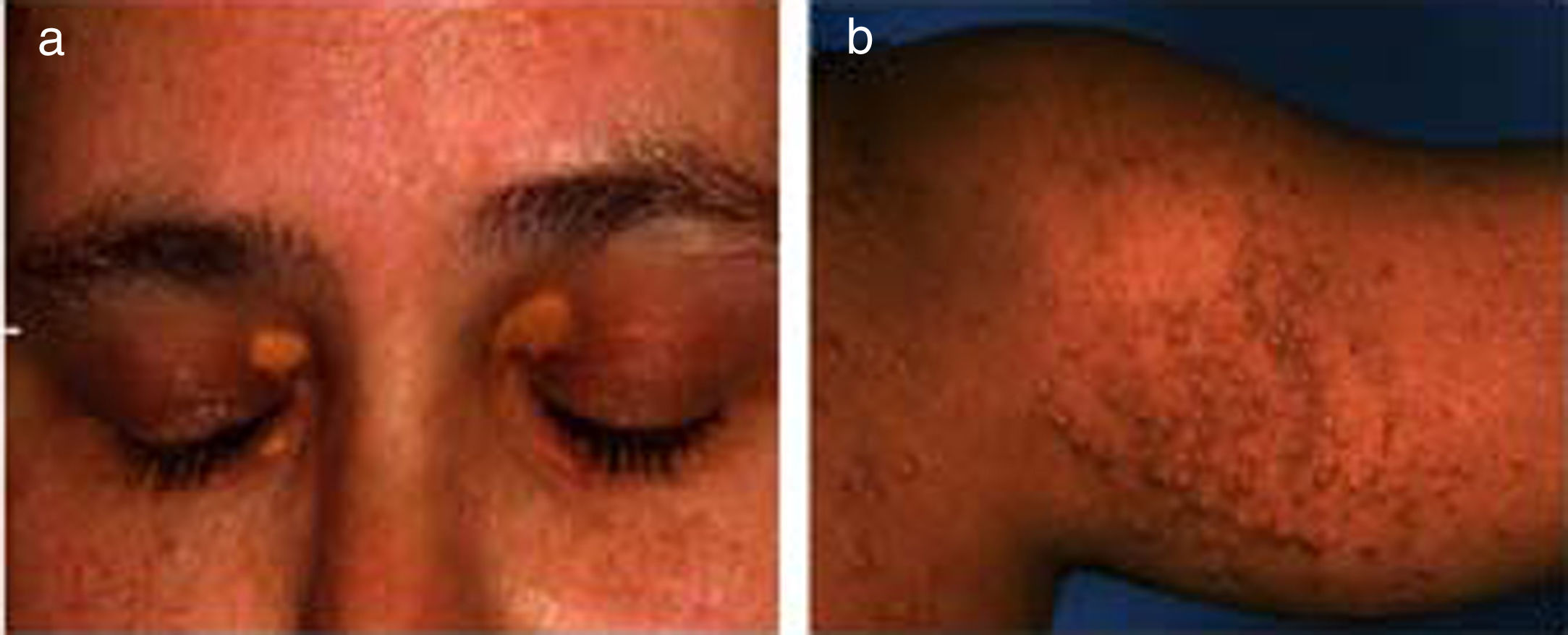
2Ocular signs in liver diseases2.1Liver disease and eyelidsXanthelasma: Orange-yellow lipid deposits of cholesterol in the skin are known as xanthelasmas [4,5]. These may be seen in patients with elevated plasma cholesterol and rarely present before the age of 16 months. Xanthelasmas are collection of lipid and cellular components characterized by soft yellow plaques, often seen bilaterally along the medial canthus of eyelids [6] (Fig. 1a). The lesions have been reported in 17% cases of primary biliary cirrhosis. On histopathological examination, they are composed of foamy histiocytes or lipid-laden macrophages [7]. However, it is not pathognomic for liver disease and can also be found in healthy individuals, in hypoproteinemia, primary biliary cholangitis, and hypercholesterolemia, etc [5–8]. Treatment is not required unless there is any visual disturbance, which occurs rarely or due to cosmetic reasons. There are many treatment options depending on patient's choice such as surgical excision, laser, or cryotherapy, however recurrence rate remains high [4]. They regress with management of hypercholesterolemia and following liver transplantation in the setting of advanced liver disease.
 Bilateral xanthelasmas of upper eyelids; (b) xanthomas involving the chest and arm.")
Xanthomas: These are depositions of yellowish, cholesterol-rich material that can appear anywhere in the body [5,8,9]. These are usually multiple, large, hard, nodulo-papular skin colored, over the extensors of joints, feet, buttocks, tendons and not just restricted to eyes [5] (Fig. 1b). It is seen along with other symptoms in many chronic liver diseases, notably Alagille's syndrome, progressive familial intrahepatic cholestasis (PFIC), and some cases of biliary atresia [5,10]. These are often seen in the conditions of hyperlipidemia and dyslipidemia as well as genetic disorders like familial hypercholesterolemia [3,4]. Disfiguring xanthomas secondary to hypercholesterolemia may accompany severe pruritus. In fact, severe intractable pruritus and disfiguring xanthomas may be a relative indication for liver transplantation in patients with PFIC and Alagille's syndrome [11]. Detection of xanthomas during childhood should prompt a thorough search for underlying systemic disease. The control of lipid profiles and underlying disorder can reverse the condition.
2.2Liver disease and conjunctivaIcterus: The conjunctiva has a strong affinity to bind bilirubin when plasma bilirubin levels rises, leading to yellowish discoloration or ‘Jaundice’. However, it becomes clinically visible only when bilirubin levels are greater than 2mg/dL [12]. The term ‘sclera icterus’ is actually a misnomer, as it is the conjunctiva and not the sclera which is involved [12]. The conjunctiva, especially the bulbar, is one of the most sensitive and early areas to detect jaundice, particularly important in patients with heavy skin pigmentation. Icterus is an indication of liver dysfunction either due to an acute insult or chronic liver disease.
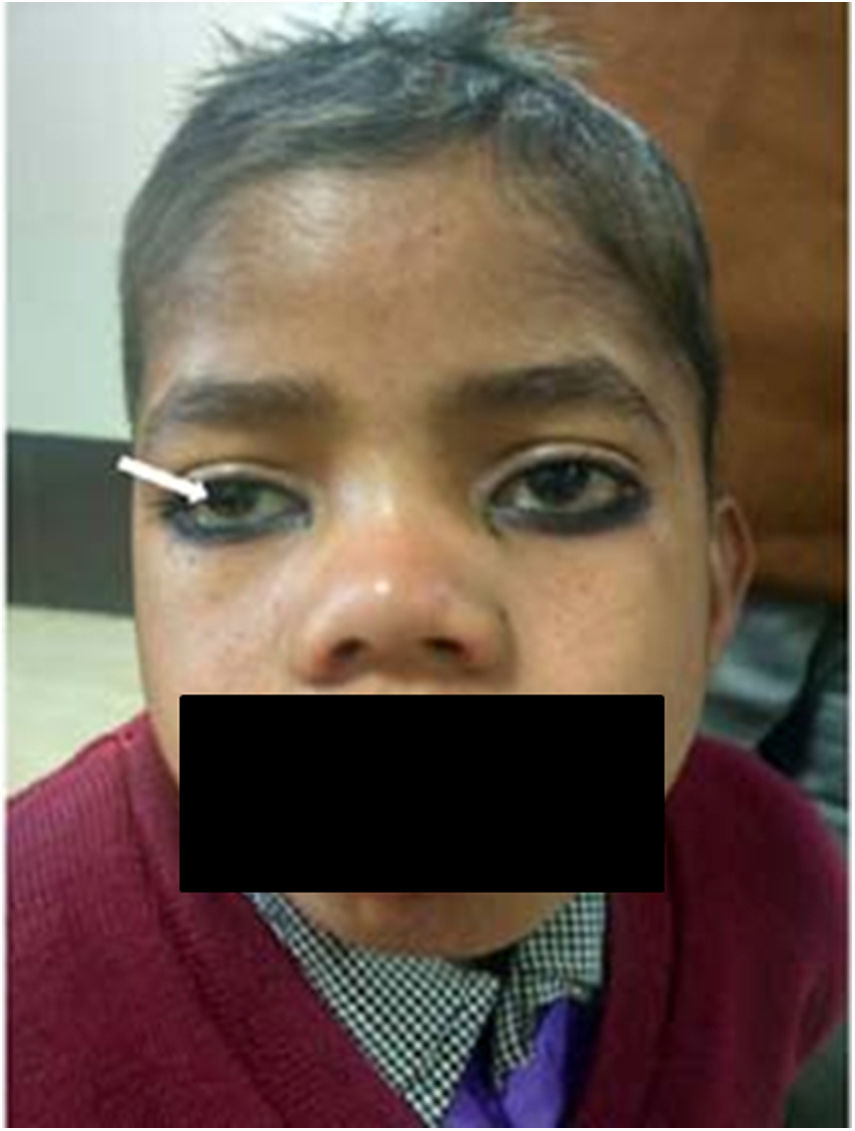
Xerophthalmia/Conjunctival xerosis: Xerophthalmia refers to a constellation of ocular symptoms and signs associated with vitamin A deficiency; which includes night blindness, conjunctival and corneal xerosis, Bitot's spot, nyctalopia, keratomalacia, corneal ulcers and retinopathy [13,14] (Fig. 2a and b). It is a major health problem in developing countries and a leading cause of preventable blindness, especially in preschool children. It is usually caused by reduced dietary intake followed by impaired intestinal absorption or defect in vitamin A metabolism and storage [14,15]. Vitamin A is a fat soluble vitamin; the main source of which is from the diet and has many systemic as well as ocular effects. The deficiency may be precipitated by malnutrition, diarrhea and respiratory tract infections in children [14]. Additionally, its deficiency is caused by fat malabsorption and chronic liver disorders [13–15]. Children with cirrhosis have an altered hepatic structure and functions which may in turn lead to Vitamin A deficient state [15]. Cirrhosis results in transformation of hepatic stellate cells (Ito cells) into activated myofibroblasts and leads to the depletion of the cells which stores vitamin A. In addition, cholestasis (deficiency or stasis of bile) which is important for fat digestion, can also lead to vitamin A malabsorption. The conditions which are known to cause cholestasis in pediatric population are PFIC, biliary atresia, bile acid synthetic defect and Alagille's syndrome, to name few [14,15]. Supplementation with Vitamin A, lubricants and treatment of underlying condition reverses the deficiency in majority. Therefore, it is justified to prescribe high oral doses of vitamin A, 5000–15000 IU daily in case of chronic liver disease and cholestasis.
 Bitot")
Bitot's spots: Bitot's spot is an early marker of avoidable blindness due to vitamin A deficiency. These spots are slightly elevated; white, foamy, triangular lesions usually seen on the bulbar conjunctiva near the limbus on the temporal side (Fig. 2a). They typically appear between 3 to 6 years of age [13,14]. The postulated pathogenesis is that Vitamin A deficiency leads to squamous metaplasia, with the cellular components in the conjunctiva becoming like rough skin rather than a mucous membrane.
Keratoconjunctivitis sicca (dry eye disease): Sjogren syndrome is a very well known cause of dry eye disease. However, dryness of conjunctiva can also be seen in other conditions like primary biliary cholangitis and chronic hepatitis C virus infection [16]. Dryness of eyes for prolonged period can result in foreign body sensation, blurred vision, redness, eyelid crusting, and potential vision-threatening epithelial erosions, corneal ulcers and infections. Thus, any patient with keratoconjunctivitis sicca should be evaluated for Sjogren syndrome as well as chronic liver disease. If symptoms persist, the condition can be treated conservatively with artificial tears but referral to an ophthalmologist is warranted.
2.3Liver disease and corneaCorneal pathology is relatively easy to detect using torch light or ophthalmoscope. However, subtle changes can only be seen by slit-lamp examination. The peripheral cornea's transparency and proximity to the limbal circulation makes it more susceptible to deposition of abnormal material from the blood. Different chemicals and substances may deposit at various levels of the cornea. The pattern, color, and depth of the deposits are often characteristic and guide the differential diagnosis. Many of the soluble chemicals may deposit in the deep stroma or at the level of the Descemet's membrane [16,17].
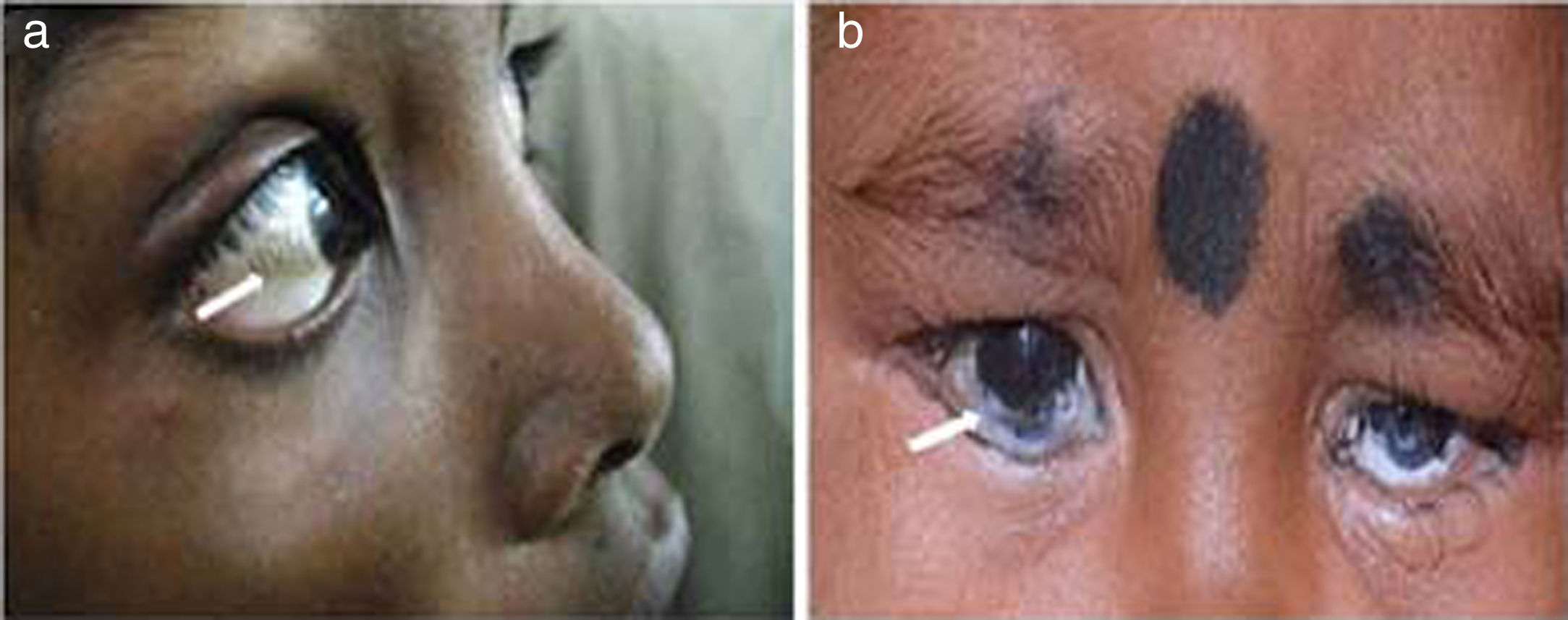
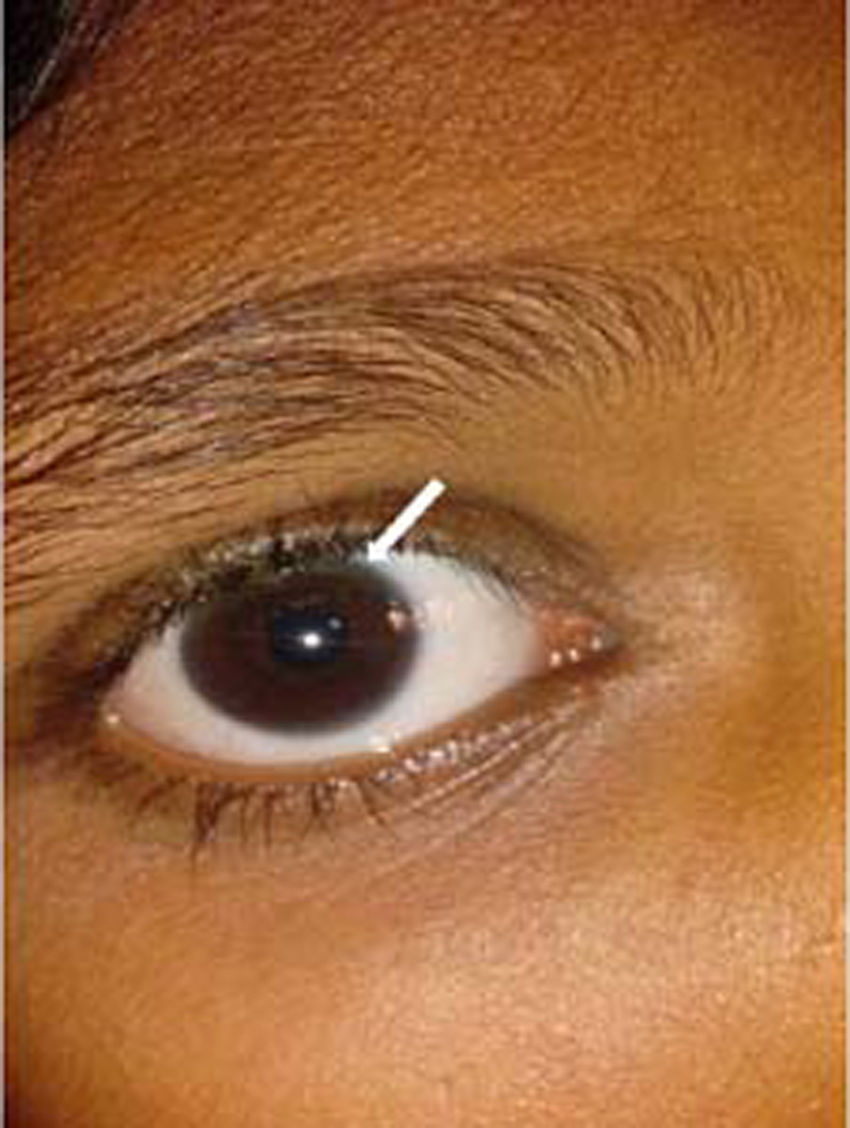
Kayser–Fleischer ring: The Kayser–Fleischer (KF) ring is a greenish-brown ring in the Descemet's membrane at the periphery of the cornea on its posteriorsurface. It is considered to be pathognomonic for Wilson's disease until proven otherwise [16,18]. It first develops superiorly in the cornea (at 12-o’clock position), then inferiorly, and finally in the horizontal meridian (Fig. 3). It is composed of granules rich in copper and sulfur. It is best detected by slit-lamp examination by an experienced ophthalmologist, but can rarely be seen by naked eye, particularly in people with blue or green pigmentation of the iris. Vision remains unaffected and there is no long-term sequelae. The pathogenic defect in Wilson's disease leads to copper overload in the body and extra copper is deposited in different parts of body, liver, cornea, brain and joints being the most commonly affected [18]. Additional ocular findings in Wilson's disease include sunflower cataracts, saccadic pursuit movements, loss of accommodation response, and apraxia of opening the eyelid [16,17]. However, cases of KF rings have also been documented in other copper related non-Wilsonian liver diseases [19,20]. It thus becomes imperative for an ophthalmologist to refer patients with a chance finding of KF ring to a hepatologist for prompt evaluation of Wilson's disease. It gradually resolves upon starting of copper-lowering therapy, which can also serve as a guide to assess the response to treatment.
.")
Pigmented corneal ring: A similar ring, called “Pseudo Kayser–Fleischer” ring can be seen in chronic cholestatic disorders including primary biliary cirrhosis owing to bilirubin deposition [19–21]. These have been reported in about 3.7% cases of primary biliary cirrhosis [19]. Advanced liver disease may cause elevation of serum bilirubin levels and yellow staining of the peripheral cornea, which is thought to diffuse from the limbal circulation. Bilirubin is found throughout the stroma layer of cornea but is more prominent in the deep stroma, in contrast to KF ring where Descemet's membrane is involved [20,21]. A cautious examination is warranted in children with deep jaundice, so as not to confuse these rings with the KF ring of Wilson's disease.
Posterior embryotoxon: Posterior embryotoxon, an abnormal prominence of Schwalbe's line (junction of Descemet's membrane with the uvea at the angle of the anterior chamber), is the most common and important ocular finding in Alagille's syndrome [22–26]. It occurs in 56–88% patients of Alagille's syndrome; however it is not pathognomonic, since it occurs in 8–15% of normal population [26,27]. A slit lamp examination therefore becomes indispensable as it cannot be visualized on naked eye examination [22]. Treatment is usually not required as it does not hamper vision. Several other ocular abnormalities like Axenfeld anomaly, Rieger anomaly, microcornea, keratoconus, congenital macular dystrophy, shallow anterior chambers, exotropia, ectopic pupil, band keratopathy, cataracts, strabismus, iris hypoplasia, choroidal folds, and anomalous optic disks have also been reported [22]. It has been hypothesized that the presence of posterior embryotoxon in setting of cholestatic jaundice may be specific for Alagille's syndrome. Additionally, children with Alagille's syndrome have a higher than normal incidence of benign intracranial hypertension and annual fundoscopy for papilledema is therefore essential [23]. Screening for posterior embryotoxon in them could aid in an early diagnosis and eliminates the need for extensive testing.
Corneal clouding: Corneal clouding is an important and specific finding in mucopolysaccharidoses (MPS) and mucolipidoses, which are inherited lysosomal enzyme deficiency disorders [1,3,28–30]. In MPS, some clinical manifestations such as coarse facial features, thickened skin, organomegaly and corneal clouding can be regarded as the direct expression of glycosaminoglycan accumulation in the tissues (Fig. 4). As a general rule, excess dermatan and keratan sulfate appears in the cornea and results in clouding. The degree of storage in the keratinocytes is related to the degree of corneal clouding. The impaired degradation of heparan sulfate is usually more closely associated with manifestations of the central nervous system and retina [28]. Among the ocular signs, corneal clouding appears within the first few years of life and is commonly seen in MPS I and IV (Hurler's and Maroteaux–Lamy syndromes). Among the mucolipidoses, it is frequently seen in type IV. [28,29]. Corneal opacities may be managed by corneal grafting (penetrating keratoplasty) if severe. Other ocular manifestations which may ultimately limit the visual acuity are the presence of retinopathy, optic nerve disease, cataract or glaucoma as well as recurrence of corneal clouding in the corneal graft [29]. It thus becomes mandatory for any child presenting with coarse facies, hepatomegaly and mental retardation to undergo an ophthalmic evaluation for corneal clouding to facilitate early recognition and referral.
.")
Corneal xerosis/ulcer/keratomalacia: We have discussed earlier that vitamin A deficiency may lead to varied ocular manifestations, affecting any part of eye from conjunctiva to retina (Fig. 2b). The deficiency usually becomes apparent when the Vitamin A stores are depleted or in conditions causing fat malabsorption. Children with cholestasis such as biliary atresia, bile acid synthetic defect, PFIC, Alagille's syndrome where fat malabsorption is common and similarly those with chronic liver disease, where vitamin A storage is limited are more prone to have vitamin A deficiency [13–15]. It is also a common occurrence in children with malnutrition, post measles infection and with frequent episodes of diarrhea [13,14]. Management consists of local cornea care with lubricants, vitamin A supplementation and treatment of underlying cause. They should be promptly referred to an ophthalmologist to prevent permanent vision loss [14].
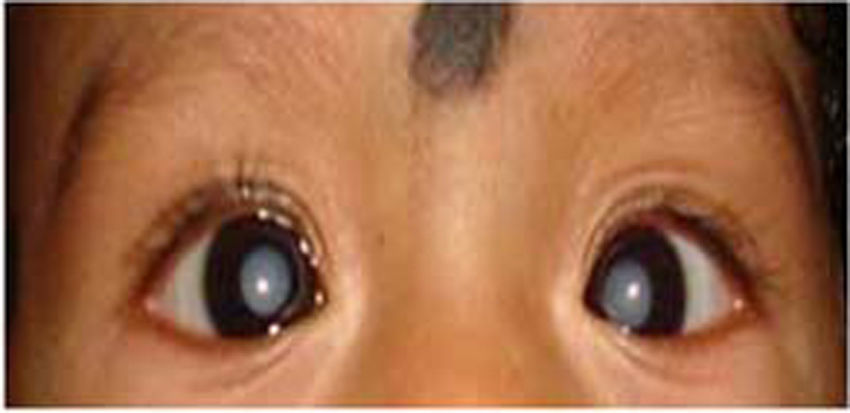
2.4Liver disease and lensCataract: The most common abnormalities of the lens are varying degrees of opacification. Various multisystem disorders have been associated with cataracts, including metabolic liver disorders [3,17]. The association of pediatric liver diseases with lens opacities is yet another important pointer for certain metabolic liver diseases. Lens metabolism is similar to that of any other tissue, with the difference being that it is avascular by nature, receiving its nutrients from the aqueous humor [17]. Galactosemia is one such disorder of galactose metabolism, which results in cataract due to the accumulation of galactitol (metabolite of galactose) (Fig. 5). Accumulated galactitol results in the shift of water into the lens, with ultimate lenticular disruption [17]. In classical galactosemia (type 1); cataract may be a manifestation which usually presents at birth or early in neonatal period along with hepatic symtoms [17,31]. The typical presentation includes vomiting, jaundice, coagulopathy or sepsis like illness and cataract. In type 2 galactosemia (galactokinase deficiency); cataract may be the sole manifestation which can present during infancy or as presenile cataract in adult patients. Naked eye examination and detection of cataract in this setting may prevent aggressive work up. Thereafter, immediate introduction of galactose free diet may reverse the metabolic derangements and manifestations. Sunflower cataract secondary to copper deposition in the eye is another ocular manifestation of Wilson's disease apart from KF ring [32,33]. The appearance of a cataract actually represents copper deposition in the lens capsule and not within the lens cortex or nucleus itself [13]. Sunflower cataracts, like KF rings, usually improve with copper-chelating drugs and have no subsequent effects on the vision. Lens membrane contains the highest cholesterol content among the known membranes, indicating the importance of normal cholesterol metabolism in lens maintenance. Thus, the diseases of inborn errors of cholesterol biosynthesis (Smith–Lemli–Opitz syndrome) and diseases of bile acid synthesis (cerebrotendinous xanthomatosis) may also be associated with cataract [17,34]. Other causes such as congenital TORCH infections, Zellweger (hepato-cerebro-renal) syndrome, neonatal adrenoleukodystrophy, neonatal hemolytic jaundice syndrome and citrin deficiency may also have lens opacities [17,33].
2.5Liver disease and retina
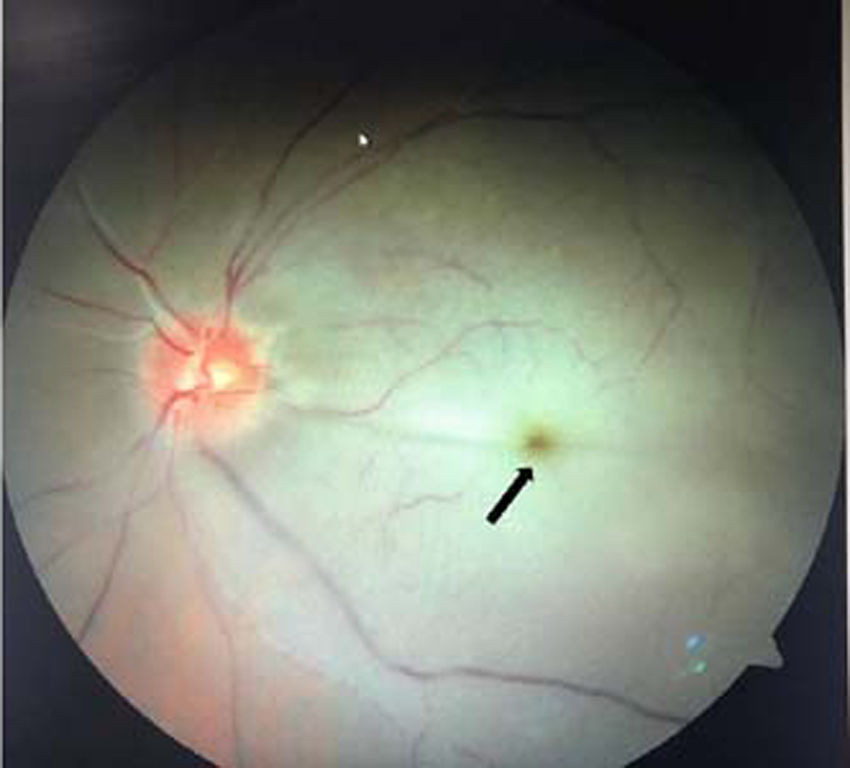
Cherry-red macular spots: A cherry-red spot is a finding in the macula of the eye seen in a variety of lipid/lysosomal storage disorders including sphingolipidoses or gangliosidoses, which includes GM1 and GM2 (Tay–Sachs) gangliosidoses, sialidosis type I and II, Fabry disease, Niemann–Pick disease, Farber disease, Gaucher disease, Krabbe disease, and metachromatic leukodystrophy [17,30,35,36]. These are grayish, peri-macular deposits of complex lipids, in which fovea remains unaffected but appears unusually red and prominent against the surrounding peri-macular infiltrates [17,30] (Fig. 6). A combination of cherry-red spot and hepatosplenomegaly is a classical feature of Gaucher's and Tay–Sachs lipid storage disease [35].

Pigmentary retinopathy and retinitis pigmentosa: Retinal pigmentary changes can be seen in various MPS disorders and are morphologically identical to retinitis pigmentosa. The MPSs exhibiting retinopathy are: Hurler's, Scheie's, Hunter's and Sanfillipo's syndromes [1,3,17,35,36]. Patients with peroxisomal disorders like Zellweger's syndrome, neonatal adrenoleukodystrophy and Refsum's disease as well as mitochondrial β-oxidation defects can also present with retinal degeneration [17,30,36]. It is also found when a mutation in a copper-transporting ATP7A gene causes Menke's disease, characterized by low serum copper levels, low ceruloplasmin levels and retinal degeneration, mental retardation, hypopigmentation and unusual kinky hair [37].
Retinal vitamin A deficiency: Chronic vitamin A deficiency and subsequent disturbance of retinal rod photoreceptor function are common in advanced primary biliary cirrhosis [14,15]. The deficiency of fat-soluble vitamins may follow chronic cholestasis due to any cause. However, this can be averted by adequate vitamin A replacement therapy [38]. Even an established night blindness may be reversed, if replacement therapy is started sufficiently early at an appropriate dosage.
2.6Liver disease and cranial nervesLiver diseases are known to be associated with several neuro-ophthalmic disorders. The manifestations may be sensory (affecting the optic nerve) or motor, which may include cranial nerve palsies, gaze palsies, internuclear ophthalmoplegia and nystagmus [17,30,33,39].
Cranial nerve palsies: These are often seen in chronic thiamine deficiency associated with poor diet and alcoholic liver disease. Wernicke's encephalopathy seen in such cases is a confusional state associated with ocular cranial nerve palsies, ataxia and nystagmus, which can also be seen in children with advanced liver disease [33]. Treatment consists of early replacement of thiamine in adequate doses which helps in reversal of the condition. Progressive external ophthalmoplegia (inability to move the eyes and eyebrows) has also been reported as one of the symptoms in mitochondrial disorder [17,33].
Gaze palsy: Dorsal midbrain syndrome (Parinaud's syndrome) has been described in Niemann–Pick disease, kernicterus and Wilson's disease [33,39]. The dorsal midbrain syndrome is characterized by vertical gaze palsy, light-near pupillary dissociation, convergence retraction nystagmus, upper eyelid retraction, skew deviation and unstable fixation. It is of great clinical significance, as vertical gaze palsy can be identified in Niemann–Pick disease when a neonate presents with liver failure or cholestasis. In addition, horizontal gaze palsy is occasionally seen in Wilson's disease [17,33,39].
Nystagmus: Nystagmus is a repetitive, involuntary eye oscillation, which can either be physiological or pathological. It is characterized by a slow phase in which the eyes deviate away from the object of fixation and then a fast, correcting phase [33,39]. It is a very non-specific sign and can be seen in hepatic encephalopathy and rarely, may also be a feature of Wilson's disease [39]. Nystagmus can be a component of hypopituitarism and may present with neonatal hepatitis and prolonged cholestasis. The diagnosis may be suspected by the presence of midline facial abnormalities (Septo-optic dysplasia), nystagmus, and microgenitalia in males [40]. Opsoclonus-myoclonus syndrome is a rare eye movement disorder characterized by ataxia (imbalance, incoordination), myoclonus (body jerks), or opsoclonus (random, multi-directional darting eyes movements), uncommonly observed in neuroblastoma as a part of paraneoplastic syndrome [41].
Optic atrophy: Mitochondrial diseases severely affect the tissues that have the greatest requirements for oxidative phosphorylation, such as the photoreceptors. Leber's hereditary optic neuropathy is a mitochondrial inherited degeneration of retinal ganglion cells. Optic atrophy has also been described in all types of MPS [17,29,30,33,39]. The cause may be multifactorial; it may be secondary to chronic papilloedema due to hydrocephalus or due to simultaneous pigmentary retinopathy [29]. It should be particularly investigated in the setting of storage disorders, when a child presents with unexplained hepatomegaly, coarse facies with or without mental retardation.
2.7Liver disease and ocular monitoringThe corticosteroids are an integral part of induction and maintenance therapy of autoimmune hepatitis, mainly due to their potent anti-inflammatory and immunosuppressive activities [42–44]. The adverse events encountered due to long-term corticosteroids therapy are still a major concern in the management. Steroids are known to induce a number of ocular side effects such as cataract, glaucoma, nonspecific keratitis, pseudo-tumor cerebri, changes in composition of aqueous and vitreous humor and variations in sclera thickness [42]. Among these, posterior subcapsular cataract and glaucoma are most commonly described in children [42]. Hence, regular ophthalmological check-up including intra-ocular pressure measurements should be done to detect raised intra-ocular pressure on time, so that corrective measures are initiated before the development of glaucoma. The dictum is ‘vision once lost, is lost forever’ and the physician should be very cautious while advising steroids and should not delay referrals to an ophthalmologist when indicated.
3ConclusionWe suggest that the diagnosis of few of liver diseases may be facilitated by ocular examination. This would allow for an early diagnosis and intervention, as in most cases, simple measures like dietary restriction and timely therapy prevents the onset of comorbidities and complications. The early recognition can facilitate both, patient care as well as genetic counselling. We therefore suggest that a detailed ophthalmological assessment should be undertaken in suspected cases where ocular signs/problems with liver diseases are known to occur.
AbbreviationsPFIC
progressive familial intrahepatic cholestasis
KF ringKayser–Fleischer ring
MPSmuscopolysaccharidoses
Source of support/fundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
ContributionsDurga Prasad: acquire and review the literature and drafted the manuscript; Arpita Bhriguvanshi: critically revised the manuscript for important intellectual contents.
Conflict of interestThe authors have no conflicts of interest to declare.



