



The coexistent of pregnancy and liver disease represent a complex clinical situation, besides the liver complications that present in pregnancy with a previous health liver, like intrahepatic cholestasis of pregnancy, acute fatty liver of pregnancy or HELLP syndrome with bleeding disorders and viral hepatitis, the previous liver damage with portal hypertension associated represent a clear stated of hemodynamic changes which increased risk of variceal bleeding. The portal hypertension syndrome has a splanchnic blood flow increase. During pregnancy an hypervolemic stated developed as consequence there is an increased in portal flow that contributed to more portal pressure transmitted to the collaterals veins which increase variceal bleeding risk in this group of patients. The present review will focus on treatment options to prevent variceal bleeding in this clinical situation.
Liver disease in pregnancy is uncommon but when it is present could be a dramatic event. There is some physiological changes that resembles pathological changes as in many uncomplicated pregnancy many laboratory tests results may appear abnormal, as serum alkaline phosphatasa increased,1 however glutamyl transpeptidase, alanine aminotransferasa, aspartate aminotranferase and bilirrubin are normally unchanged.2 Liver dysfunction during pregnancy can be caused by conditions that are specific to pregnancy or by liver disease that are not related to pregnancy. The most common hepatocelular liver diseases in pregnancy are: intrahepatic cholestasis of pregnancy, acute fatty liver of pregnancy, preeclampsia and eclampsia, HELLP syndrome with bleeding disorders and viral hepatitis. Preeclampsia is characterized by a triad of hypertension, proteinuria and edema where hypertension is defined as an elevation of 30 mmHg (systolic) or 15 mmHg (diastolic) above the value in the first trimester or any value above 140/90 mmHg. Eclampsia is marked by seizures or coma in addition to preeclampsia.3,4 The HELLP syndrome (Hemolysis, elevated liver enzymes and low platelets) is a complication of severe preeclampsia. Weinstein originally described this condition in 29 patients and used the acronym of HELLP5 but Pritchard in 19546 recognized the association of hemolysis, elevated liver enzymes and thrombocytopenia. Women with the HELLP syndrome presents with right upper quadrant or epigastric pain (65-90%), nausea or vomiting (36-50%), headache (30%). Jaundice presents in only 5%. Physical examination revels right upper quadrant tenderness (80%), weigh increases with edema (60%) and hypertension could be absent in 20%.
The frequency of HELLP syndrome is variable: 0.1% to 0.6% of all pregnancy and 4% to 12% of preeclamptic women.7 Most of cases, seventy percent, presents between 27th and 36th week of gestation, and the other third occur post partum. The typical laboratories abnormalities are: microangiopathic hemolytic anemia, elevated serum lactate dehydrogenase concentrations, aminotransferase concentrations 2 to 10 fold increases and low platelets < 100,000/mm3. The basic cause of preeclampsia-eclampsia is unknown. Epidemiologic studies suggest an immunologic cause for preclampsia, since it occurs predominantly in women who have had minimal exposure to sperm (having used barrier methods of contraception) or have new consorts, in primigravidas, and in women both of whose parents have similar HLA antigens. Preeclampsia is an endothelial disorder resulting from poor placental perfusion, which releases a factor that injures the endothelium, causing activation of coagulation and an increased sensitivity to pressors, in fact, before a florid clinical manifestation, there has been demonstrated vasospasm in small vessels, accounting for the pathologic changes in the placenta with consequent adverse effects on the fetus. As one could expect pathogenesis of HELLP syndrome has not been fully understood, although mechanisms similar to preeclampsia have been proposed. As in preeclampsia, the mechanism may be attributed to activation of the complement and coagulation cascades, increased vascular tone, platelet aggregation, and alteration of the tromboxane: prostacyclin ratio, leading to systemic endothelial and microvascular injury and causing microangiopathic hemolytic anemia, periportal hemorrhage, periportal hepatic necrosis and thrombocytopenia.
The peripheral blood smear shows microangiopathic hemolytic anemia. The liver histology revels periportal hemorrhage and periportal or focal parenchyma necrosis with hyaline deposits. At sinusoidal level fibrin microthrombi and fibrinogen deposition may occur. Steatosis occurs in one third of patients. There has been reported hepatic infarction and the liver capsule occasionally can rupture from an underlying hematoma.
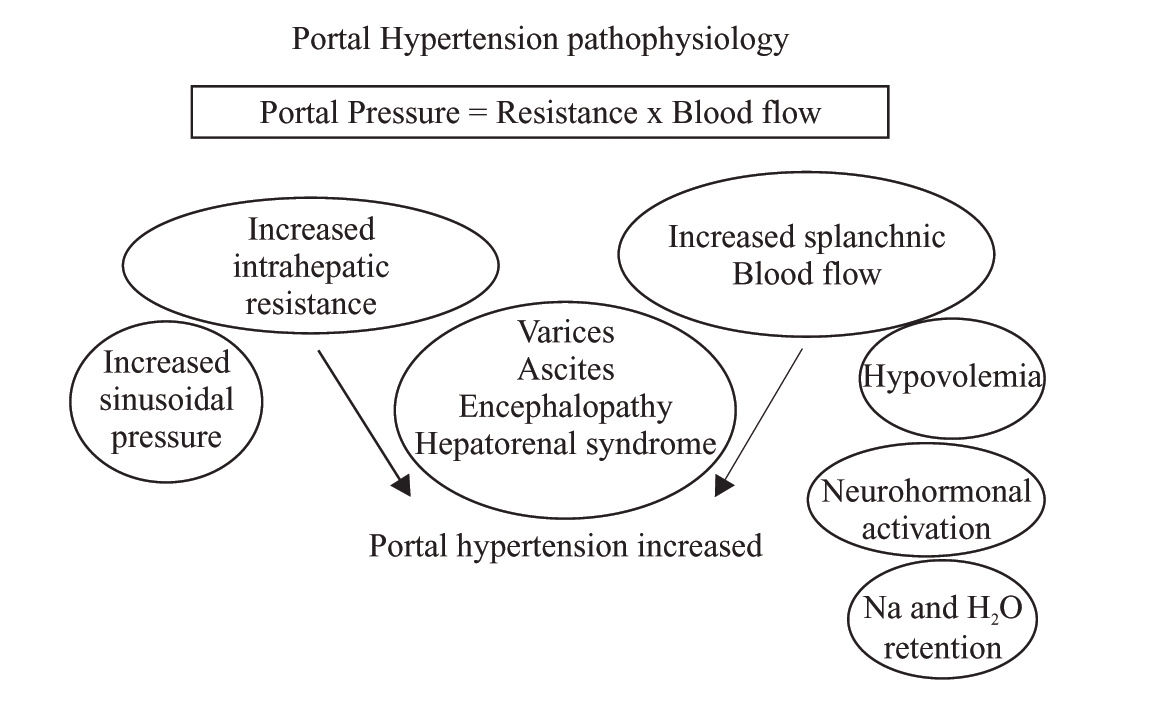
Portal hypertension profile in pregnancyBesides all this catastrophic complications in preclampia-eclampsia, there is another context in pregnancy related to the portal hypertension syndrome, of course that pregnancy is rare in the context of portal hypertension in liver disease woman, but in fact both could coexist and both situations share hemodynamic changes: Pregnancy modify systemic hemodynamics early in the first trimester in response to increased oxygen requirements both for the fetus and for the mother, as consequence plasma volume increased since the sixth to 32th week of pregnancy with a global increased of 50%, (Figure 1).8 This volume increased is directly related with an increased in sodium retention resulting in a global retention of 1,000 mEq of sodium. Increased tubular reabsorption is generated by increased plasma aldosterona levels and increased in estrogen, deoxycortisone and placental lactogen levels. The reninangiotensin levels changes with an up tenfold increased, as is described below, all this hemodynamics changes are also present in portal hypertension. Pregnancy produces water retention beyond that expected from the sodium retention and this could be related to activation of specific water channels (aquaporinas) in distal nephron, so water move from hypo-os-molar to hyperosmolar interstitium. Both sodium and water retention contributes to volume expansion, as volume increased, witch means sodium and water retention, also erythrocyte mass increased by 30%, although plasma volume increase more than erythrocyte mass with a significant decreased in maternal hematocrit. Maternal cardiac output increased by 30% to 50% during pregnancy. In the other side, circulatory function is severely impaired in cirrhotic patients due to a splanchnic arterial vasodilatation.9 An increased local release of nitric oxide and other vasodilators related to portal hypertension is the most likely explanation of this abnormality.10 Compensatory mechanisms, such as an increased cardiac output and overactivity of the renin-angiotensin system, sympathetic nervous system and antidiuretic hormone, are essential in the maintenance of arterial pressure in cirrhotic patient, Figure 1 unfortunately this mechanisms are insufficient and cirrhotic patients develop marked hemodynamic disturbances know as hyperdinamic syndrome with high cardiac output, low mean arterial pressure and low systemic vascular resistances. Although, the circulation of these patients is expanded and hyperdynamic, they are from a functional point of view hypovolaemic, thus they have enhanced sympathetic nervous activity and activated renin-angiotensin-aldosterone system.11 All this hemodynamics changes are well described in cirrhotic patients with sinusoidal portal hypertension and in patients with portal venous obstruction (prehepatic)12 but it was unknowed if postsinusoidal (posthepatic) portal hypertension (Budd Chiari Syndrome) shared the same hemodynamics abnormalities, recently we described the hemodynamics and plasma volume changes in Budd Chiari Syndrome patients13 we showed that these patients had activation of vasoactive neurohumoral systems and expanded plasma volume, this was observed even though most of the patients did not exhibit systemic vasodilatation and cardiac output was not increased in contrast with what is observed en cirrhotic patients. In the physiopathology of portal hypertension there is an increase in portal blood flow due to splanchnic vasodilatation. Moreover there is also an intrahepatic resistance to this blood flow, which is the first known mechanism of portal hypertension. This is not only due to an alteration in hepatic architecture but also to a dynamic situation originated by the contraction of perivascular smooth muscle cells, myofibroblasts and hepatic stellate cells that represent approximately 30% of the global intrahepatic resistance. This concept was first described by Bathal and Groszmann.14 The aforementioned mechanisms increase pressure in portal territory and, by using the hepatic venous pressure gradient (HVPG = wedge hepatic pressure - free hepatic pressure) as a reflex of the portal pressure, is known that it is required a HVPG > 10 mmHg for esophageal varices to appear and over 12 mmHg for those varices to bleed.15 The hepatic venous pressure gradient is the gold standard to measure the portal pressure to guide therapy and prognosis in cirrhotic patients who have previously bled, the portal pressure measurement is not a new technique, in 1937 Thompson published the first results of portal pressure measurements in humans direct to the portal vein. In 1951 Myers and Taylor developed the technique of hepatic venous pressure as surrogate of portal pressure:16 A catheter is guided under fluoroscopic control to the hepatic vein through transjugular access and measure wedge hepatic pressure (WHP) which reflects sinusoidal pressure which equal portal pressure in liver disease. The catheter is pulled out to register the free hepatic pressure (FHP). The gradient between wedge and free hepatic pressure represents the hepatic venous pressure gradient (HVPG=WHP-FHP). At the beginning there were troubles about the reproducibility of measurements, there were a lot of variations, so the reproducibility was low. To solve this situation in the 60’ years, the balloon tip catheter was introduced with some advances: the balloon occludes the hepatic venous flow in a large territory so the sinusoidal pressure is recorded in a larger area than the conventional catheter. It is recommended to take the pressure three times and to use slow recording speed for accurate interpretation of the tracing.

Portal hypertension is definite as the pathologic increase of portal pressure expressed as HVPG, normal values are considerate between 1-5 mmHg.17
The clinical manifestations of the portal hypertension syndrome are: Variceal bleeding, ascites, encephalopathy and hepatorenal syndrome. More than 40% of patients with cirrhosis have esophageal varices at the moment of the diagnosis. About 30% of those with large varices will have a bleeding episode in the next two years18 with a one-year rebleeding possibility of about 60% and a mortality of 20% in each episode.19 There are few data about the effects of cirrhosis on pregnancy, cirrhotic patients have a high incidence of fetal wastage from 10% to 66%20,21 and also have a spontaneous abortion rate of 20%, but patients with non cirrhotic portal hypertension like portal vein thrombosis non associated to cirrhosis have the same rate of spontaneous abortion that the general population estimated from 3% to 6% which means that this subgroup of patients have portal hypertension but with a preserved liver function, in fact this same group of patients have better fertility than the cirrhotic patients. Perinatal mortality is increased in patients with portal hypertension, 11% to 18%, some series from the India have described higher mortality: 33% associated with variceal bleeding during pregnancy. As one could expect, portal hypertension associated complications occur in 30% to 50% of pregnancy in presence of portal hypertension. Toxemia of pregnancy is not more frequent in presence of portal hypertension syndrome. The most catastrophic event is variceal bleeding, 75% of patients with varices will bleed during pregnancy,22,23 more than twice than the expected bleeding rate in cirrhotic patients with large varices (30% in the next 24 months), besides mortality variceal bleeding associated ranges from 18% to 60% in cirrhosis and pregnancy whereas the mortality in non cirrhotic patients is < 10%.24 The approach of pregnancy and portal hypertension should included three different scenarios: Preconception, prenatal and perinatal. Young women with cirrhosis or non cirrhotic portal hypertension should counseling about pregnancy risks related to all the complications describe above, besides the risks of transmitting hepatic disease to the newborn, as in the case of viral cirrhosis. Pregnancy could be planned only in cases of stable liver disease in absence of complications, if this late situation is absent, contraception should be recommended. If esophageal varices are present the risk of bleeding should be evaluate, as the past history of bleeding from variceal source. The treatment options are: Propranolol and nadolol, which are non-selective beta blockers, reduce portal pressure via two mechanisms: 1) Cardiac output is reduced by blocking b1 adrenergic receptors, 2) Splachnic vasoconstriction by blocking b2 receptors (vasodilators). By these two mechanisms splanchnic flow is reduced, as is portal pressure, reflecting a reduction of pressure in collateral veins (varices) and also in their walls.
The usefulness of beta blockers has been evaluated and compared against placebo in 12 randomized trials. Several meta-analyses show a reduction in bleeding risk, demonstrating that non-selective beta blockers constitute an efficacious therapy in primary prophylaxis. Patients with large varices had a 30% risk of first bleeding in the following 24 months; beta blockers reduced this risk to 15%.25,26 This means that beta blocker utilization allows a global reduction of 50% in risk of first variceal bleeding episode. It is clear that nonselective beta blockers do not protect all patients, because there remains a 15% bleeding risk in the subsequent 2 years in patients using beta blockers; nonetheless, this fact might be due to insufficient reduction in HVPG and to differences in individual sensitivity to beta blockers depending on age, weight, genetic polymorphisms of beta adrenoreceptors, and amount of portosystemic collaterals. According to these facts, treatment of 11 patients is required to prevent one variceal bleeding episode.
Previous reports demonstrated that reducing HVPG to less than 12 mmHg nearly totally lowers risk of bleeding.27 Other studies have shown that even without reaching these values, reducing HVPG by at least 20% of the basal value is related with lower bleeding risk, estimated between 4 and 9% in 1 and 2 years, respectively.28 Also, it has been demonstrated that patients reaching the previously mentioned hemodynamic goals exhibit a marked reduction in the risk of developing other complications of portal hypertension such as ascites, spontaneous bacterial peritonitis, hepatorenal syndrome, and hepatic encephalopathy, in addition to having improvement in survival rates when compared with non-responders.29
Once the hemodynamic goal is reached, it is sustained in the majority of patients.30 In primary prophylaxis, endoscopic sclerotherapy is not recommended due to the morbidity related with this procedure. A meta-analysis comparing endoscopic variceal ligation (EVL) vs. beta blocker treatment31 estimated an odds ratio (OR) for first bleeding episode of 0.48 (0.24 to 0.96), with a necessary number to treat (NNT) of 13 patients in favour of EVL, although it does not offer great advantages with regard to survival. Thus, EVL is not recommended as primary prophylaxis32 at present EVL should be considered for the patient who is unable to tolerate or does not respond to beta blocker therapy.
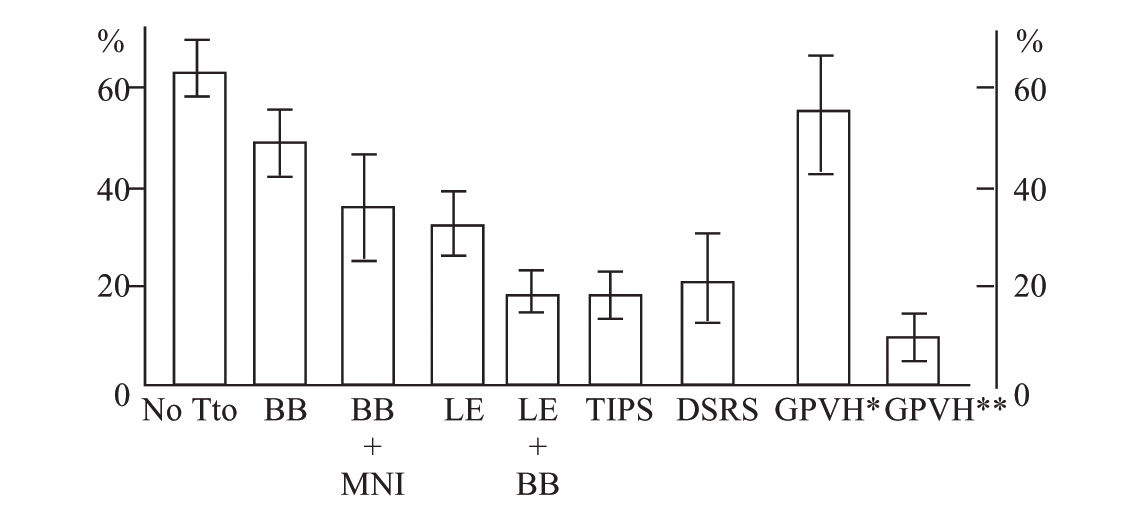
Baveno’s consensus32 highlighted endoscopic banding as a first-line treatment, similar to beta blocker therapy in secondary prophylaxis (rebleeding prevention). There are three trials that compare pharmacologic treatment (beta blocker plus isosorbide mononitrate) vs banding. One shows more benefit from the pharmacologic therapy,33 the second shows an identical benefit from both treatments,34 and the third one places endoscopic banding over pharmacologic therapy,35 without a difference in mortality in any of these. On the other hand, older trials that compared re-bleeding frequency between both treatments found that banding had a re-bleeding frequency of 16 to 29%; more recent trials found a higher re-bleeding frequency (38 to 56%).36 This finally states that endoscopic banding is not better than beta blocker treatment. In addition, banding acts locally in varices without improving portal hypertension physiopathology. Endoscopic banding is effective, but for a short time only because portal pressure and flow are not modified and there is a recurrence of varices up to 50% in 2 years. This renders endoscopic follow-up necessary.37 The combination of beta blocker and ligation to prevent the rebleeding could be better than ligation alone38,39 with a low frequency of rebleeding with combining banding and beta blocker vs. banding alone (23 vs 47%, respectively; p = 0.005). Suggesting that the combination of banding and beta blocker can be a useful alternative. Figure 2 shows different treatment options in the prevention of variceal rebleeding. The principal risks of beta blocker therapy are fetal bradycardia and growth retardation which could make the band ligation a better option during pregnancy. At present, surgery and TIPS are only recommended as rescue therapies in patients with failure in endoscopic or pharmacologic treatments. During pregnancy the phycian must consider benefits over the risk of each one of the therapeutic options describes above to prevent variceal bleeding or rebleeding in the case of past haemorrhage. There are no control trails to overemphasizes one treatment over the other in pregnancy and most of the recommendations come from cirrhosis trials, finally there is not know about the best way of delivery, but in fact one must be care about volume overload, coagulation disorders and sedation administered.




