







Background and aim. Hepatocellular carcinoma (HCC) is the most common primary liver cancer diagnosed worldwide. Deregulation of Wnt/β-catenin pathway has been associated with the development of HCC in a substantial number of cases in Europe and far less in Asia. Nothing is known about this pathway in HCC cases from South America. This study aimed to investigate the frequency of mutations in β-catenin gene (CTNNB1) and the subcellular localization of β-catenin in HCC cases from Colombia.
Material and methods. We determine by direct sequencing the frequency of mutations in exon 3 of CTNNB1 gene and by immunohistochemistry the subcellular localization of β-catenin in 54 samples of HCC obtained from three pathology units in Bogota and Medellin cities.
Results. Only three HCC cases (5.6%) were found mutated at residues (G34E, S45P, P44S, T41I) important for phosphorylation and ubiquitination of β-catenin protein. Strikingly, nuclear or cytoplasmic accumulation of β-catenin, hallmark of Wnt pathway activation, was found in 42.6% HCC cases (23/54). Interestingly, β-catenin accumulation was significantly more frequent in young patients and hepatitis B virus-related HCC.
Conclusions. Although, CTNNB1 exon 3 mutations are not frequent in HCC from Colombian patients, our findings indicate that Wnt/β-catenin signaling is activated in 42.6% of HCC samples. Furthermore, Wnt signaling was demonstrated in HCC cases associated of HBV infection, one of the most important HCC risk factors in Colombia.
Primary liver cancer is one of the most frequent malignant tumors and the third cause of cancer mortality worldwide. Hepatocellular carcinoma (HCC) represents the most common primary liver cancer. Around 80% of HCC cases are related with chronic liver disease with development of fibrosis and cirrhosis. Indeed, the principal HCC risk factors are chronic infections with Hepatitis B virus (HBV) and Hepatitis C Virus (HCV) and alcohol liver disease. Dietary exposure to Aflatoxin B (AFB), hereditary hemochromatosis and non-alcoholic steatohepatitis are also HCC risk factors.1–3
Several genetic and epigenetic changes and deregulation of signaling pathways have been described in HCC; this heterogeneity could be related to the different risk factors and the host genetic background. However, the principal molecular cellular pathways implicated in liver carcinogenesis regardless the etiologies are Wnt/β-catenin, Hepatocyte growth factor (HGF)/Met, Epidermal growth factor receptor (EGFR)/RAS/MAPK and PI3K/AKT.4–6
The Wnt/β-catenin pathway is highly conserved through evolution and is critical on different cellular process such as proliferation, morphology, differentiation, organ development and survival. β-Catenin, encoding by CTNNB1 gene, is the central component of Wnt pathway. However, this protein was originally described by its interaction with the cytoplasmic domain of cadherin and α-catenin and its key role in cell-cell adhesion.7,8 Indeed, β-catenin is essential for the adherents junctions in epithelial cells; disruption of the interaction among β-catenin, cadherin and actin cytoskeleton is important for development and turnover of tissues, but also for motility and metastasis in aberrant growth.9
The Wnt Signaling pathway is activated by the binding of a Wnt ligand to the membrane receptor Frizzled (Fz) and to the low density lipoprotein receptor-related protein 5/6 (LRP5/6). In consequence, the cytoplasmic destruction complex is not assembled resulting on accumulation of stabilized β-catenin in the cytoplasm. This complex is consisting of serine-threonine kinase-I alpha (CK1α), glycogen synthase kinase 3 β (GSK3-β), the scaffolding protein, Axin and Adenomatous polyposis coli (APC); recently, it was demonstrated that Wnt signaling inhibits GSK3-β activity by sequestration of this kinase in multivesicular endosomes.10
β-catenin is translocated from the cytoplasm to the nucleus where functions as a transcriptional activator in conjunction with Tcf/Lef (Lymphoid enchancer factor/Tcell factor) DNA binding proteins. The most important target genes of this pathway are c-myc, Cyclin D1, c-jun, vascular endothelial growth factor (VEGF), matrix metalloproteinase-7 (MMP-7) and CD44.7,11 Additionally, the target genes in the liver include glutamine synthetase, cytochrome P450, EGFR, leukocyte cell-derived chemotaxin 2, senescent marker protein-30 and Epithelial cell adhesion/activating molecule (EpCAM).12
In the absence of the Wnt ligand, cytoplasmic β-catenin is constantly phosphorylated by the kinases CK1α and GSK3-β, as components of the degradation complex (Axin, APC, CK1α, GSK3-β). Phosphorylated β-catenin is ubiquitinated and therefore become a target for the proteasome degradation maintaining free cytoplasmic β-catenin at a low level. Subsequently, Wnt target genes are repressed.
Mutations in CTNNB1 gene are described in numerous human cancers, including HCC. These mutations that change residues target for phosphorylation (S33, T41, S45) or residues adjacent to them, abrogate the phosphorylation of the β-catenin protein and in consequence its degradation; thereby generating the stabilization of cytoplasmic β-catenin and subsequently translocation to the nucleus and triggering the transcription of Wnt target genes relevant to cell proliferation and apoptosis.13–15
Deregulation of Wnt pathway has been described in 17-40% of HCC cases due to different mechanisms including mutations in CTNNB1, Axin-1 and Axin-2 genes, inactivation of GSK3-β and promoter hypermethylation of genes coding for components of this signaling cascade.16–18
In the current study we examined HCC cases for CTNNB1 exon 3 mutations and for subcellular localization of β-catenin protein. The 54 HCC cases were collected in three reference pathology units in Colombia.
Material and MethodsLiver samplesHCC samples obtained from 54 patients were retrospectively studied (32.6% female, 67.4% in male). The specimens were registered at the Pathology Units of Fundación Santa Fe de Bogotá (FSB), Pablo Tobón Uribe Hospital (HPTU), and Faculty of Medicine, University of Antioquia (UdeA). These pathology units corresponded to national leading institutions and additionally two of the most important hospitals in Medellin (HPTU) and Bogota (FSB) cities.
Two independent pathologists assigned the histological pattern and grade of tumor differentiation. The HCC cases included in this study were classified according to the Edmonson-Steiner criteria as G1 well differentiated (5.6%), G2 moderately differentiated (72.2%), G3 poorly differentiated (18.5%) and G4 undifferentiated (3.7%). The trabecular type was the most frequent growth pattern (59%) followed by solid (19%), acinar or pseudo-glandular (13%) and mixt (9%).
In a previous study, HBV biomarker was demonstrated in 12 from 20 HCC samples (60%) analyzed by HBx protein immunohistochemistry detection and/or HBx sequence PCR detection; while the HCV Core protein was detected by immunohistochemistry in 8 from19 HCC cases (42%).
The TP53 mutation at codon 249 (249ser) was not detected in 21 HCC samples analyzed for this biomarker (AFB1 exposition); although, A12708G/ R213R (exon 6), G12457T/V157F (exon 5) and G13804A/C275Y (exon 8) silent and missense mutations were present in 3/21 HCC samples.19
Ethics statementAll procedures adopted in this study were followed the terms established by the Ethic Committee of Universidad de Antioquia (N.125, SIU, Universidad de Antioquia).
β-catenin gene mutations analysisGenomic DNA was extracted from tumor tissue areas of unstained paraffin sections. Briefly, the sections of 4 µm were deparaffined in xylene and ethanol and afterward the area of interest was scrapped into 1.5 mL sterile microcentrifuge. DNA was obtained using QIAmp DNA Micro kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. DNA extracts were stored at −20 °C.
Mutation analysis of the CTNNB1 gene (exon 3) was performed on HCC samples by two PCR strategies using the primers BetaGF (CCAATCTACTAATGCTAATACTG), BetaGR (CTGCATTCTGACTTTCAGTAAGG), InbetaCF (CTGATTTGATGG AGTTGGAC) and BetaCR (TACTCTTACCAGCTACTTG) previously described.15,20
The first strategy was a touch-down PCR (TD-PCR) using the BetaGF and BetaGR primers, followed by a hemi-nested PCR using InbetaCF and BetaGR primers. The second strategy was a TD-PCR using BetaGF and BetaCR primers and a hemi nested PCR using InbetaCF and BetaCR primers.
The PCR were carried out using 5 of DNA, 0.5 U of Platinum® Taq DNA polymerase High Fidelity (Invitrogen Carlsbad, USA) and 0.3 μΜ of each primer. Cycle conditions of the first strategy were: initial step of denaturation at 95 °C for 2 min, cycling steps (x20) of denaturation at 95 °C for 30 s, annealing temperature starting at 69 °C for 30 s (decreasing by 1 °C/2 cycles) and extension at 72 °C for 1 min. Followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 59 °C for 30 s, extension at 72 °C for 1 min and final extension at 72 °C for 10 min. The profile of the hemi-nested PCR of the first strategy was initial denaturation at 94 °C for 2 min, cycling steps (x 30) of denaturation at 94 °C for 15 s, annealing at 46 °C for 30 s, extension at 72 °C for 30 s and final extension at 72 °C for 5min.
The conditions of the second TD-PCR strategy were similar to the conditions of the first one, except for the starting annealing temperature (59 °C decreasing by 1 °C/2 cycles) and the annealing temperature of the hemi-nested PCR (50 °C). The PCR products were checked on standard agarose gel (First strategy 273-pb fragment and Second Strategy 249-pb fragment).
Finally, the PCR products were purified and sequenced (Macrogen, Korea) using the automated dideoxynucleotide method (BigDyeTM terminator). The mutations were verified in both sense and antisense directions.
Immunohistochemical analysis of β-catenin proteinAnalysis of β-catenin expression was performed on serial sections (4 μm) from each tumor. The section were deparaffinized and rehydrated. Then, antigen retrieval was performed using citrate buffer (pH 6.0) (DAKO, USA). The endogenous peroxidase activity was blocked by hydrogen peroxide (6%) for 5 min.
Staining was performed using mouse monoclonal anti-human β-catenin (DAKO, USA) as primary antibody at a 1:200 dilution and the kit ultravision LP detection System HRP Polymer & DAB Plus Chromogen (Lab Vision Corporation).
The pattern expression was evaluated in the HCC samples as well as in positive (hepatoblastoma) and negative (Normal Liver) tissue controls in each assay. A semi quantitative scale was used to assess the degree of positivity in ten 40x fields: + 1 to 25% positive tumor cells, ++ 26 to 50%, + + + 51 to 75% and ++++ 76 to 100%.
Additionally, subcellular immunostaining of β-catenin was categorized as nuclear (N) in samples with nuclear with or without cytoplasmic staining. HCC cases with significant cytoplasmic staining (+++/ ++++) without nuclear staining were classified as cytoplasmic (C). Finally, the HCC cases displaying membranous with or without some cytoplasmic staining was categorized as membrane (M).
Statistical analysisCategorical data were compared using chi-square test. All statistically analysis were performed using SPSS version 17.0 (SPSS Inc., Chicago IL), and p value < 0.05 was considered statistically significant. Class discovery was performed by hierarchical clustering using the dChip v.2010 software.21
ResultsDemography and baseline characteristicsThe mean age of patients was 65± 8 years (range 48-88, IQ 59-71 years) and the sex ratio (Males:Females) was 2.3. The age of female patients was significantly higher than those of males (70 ± 9 vs. 62 ± 4, p = 0.004). The bases of such difference were unknown as both HBV and HCV biomarkers prevalence were similar in the two sexes. We suspect, however, that alcohol consumption could play a crucial role in the anticipation of liver tumor development in men. Histological pattern analysis indicated that trabecular type was dominant in this series (59% of cases), followed by solid type (17%), acinar/pseudo-acinar (14.5%) and mixt (9%). Regarding tumor differentiation, 78% of tumors were defined as grade I or II and the remaining as grades III-IV (22%) (Table 1).
Demographic data, tumor atributes and biomarkers of HCC cases from Colombia.
| Clinico-demographic features | n(%) | P value |
|---|---|---|
| Sex | ||
| Men | 35(70 | |
| Women | 15(30) | |
| Sex Ratio M:F | 2.33 | |
| Age (mean) | 65 ± 8 | |
| Womens age | 70 ± 9 | 0.004 |
| Mens age | 62 ± 4 | |
| Origin | ||
| Bogota | 21(39) | |
| Medellin | 33(61) | |
| Histological pattern | ||
| Trabecular | 32(59.0) | |
| Solid | 9(17.0) | |
| Acinar/Pseudo-acinar | 8(14.5) | |
| Mixt | 5 (9.0) | |
| Edmonson-Steiner grade | ||
| I | 3(5) | |
| II | 39(72) | |
| III | 11(21) | |
| IV | 1(2) | |
| Viral biomarkers (n = 20) | ||
| HBV+ (HBsAg, and/or HBVDNA) | 12(60) | |
| HCV+ (anti-HCV, HCV RNA) | 8(40) |
The 54 HCC samples were screened for mutations in CTNNB1 gene exon 3 encoding for the β-catenin protein domain target of GSK-3 β. Using the first TD-PCR strategy the fragment was amplified in 33 HCC samples while in the others 21 HCC samples it was amplified using the second TD-PCR strategy.
Four CTNNB1 gene mutations were identified in 3/54 (5.6%) HCC cases. One mutation was detected on the threonine residue target of the GSK-3 β phosphorylation (T41I), two mutations at sites for CK1a binding (P44S, S45P) and the fourth mutation at the contiguous residue of serine 33 (G34E).
The mutation S45P (30195T > C) was detected in one HCC case (sample #40), a transition that change the residue of serine to proline; additionally this case revealed the mutation P44S (30192C > T), a transition that also modify a residue of serine to proline. Another mutation on the phosphorylation sites was identified in one HCC case (sample #61). A transition 30184C > T at codon 41could modifies the amino acid threonine to isoleucine (T41I). These mutations modified three important phosphorylation sites for β-catenin protein and consequently for its degradation.
The last mutation detected in one case (Sample #23) revealed a G to A transversion at codon 34; this mutation G34E showed an amino acid changed of glutamic acid to glycine that could modify a ubiquitination-target motif (Figure 1).
. A. Sequence from HCC case wild type. B. Sequence from case #23 HCC with mutation G34K. C. Sequence from case #40 HCC with mutations S45P and P44S. D. Sequence from case #61 HCC with mutation T41I. F Seq: Forward sequence. R Seq: reverse sequence.")
CTNNB1 mutations in HCC cases from Colombia. Representatives sequence chromatogram of CTTNB1 gene (codon 32 to 47). A. Sequence from HCC case wild type. B. Sequence from case #23 HCC with mutation G34K. C. Sequence from case #40 HCC with mutations S45P and P44S. D. Sequence from case #61 HCC with mutation T41I. F Seq: Forward sequence. R Seq: reverse sequence.
Subcellular localization and level of expression of β-catenin protein was examined by immunochemistry in HCC cases (Figure 2). Nuclear accumulation of β-catenin (+ to +++) with or without cytoplasmic staining was detected in 10 HCC cases (18.5%); this subcellular localization represented the activation of Wnt/β-catenin signaling. However, no mutations at exon 3 of CTNNB1 gene were observed in these samples.

β-catenin expression in HCC cases from Colombia. Representative immunochemistry of HCC cases. A. Positive control from Hepatoblastoma case with nuclear β-catenin expression. B. Cytoplasmic β-catenin expression in a liver sample without HCC. C. Nuclear β-catenin expression in a HCC case without mutations in CTNNB1 gene. D. Nuclear and Cytoplasmic β-catenin expression in a HCC case with mutation in CTNNB1 gene.
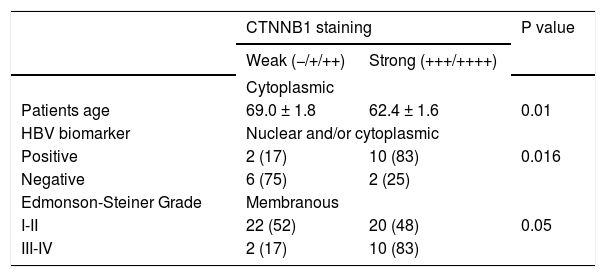
Thirteen HCC cases (24.1%) showed significant cytoplasmic staining (≥ + + +) without nuclear staining. Interestingly, patients with a strong cytoplasmic accumulation of β-catenin were significantly younger than those with low staining (62.4 ± 1.6 vs. 69.0 ± 1.8, p = 0.0161). These samples revealed transient activation of the pathway, including two of the samples with CTNNB1 exon 3 mutations (#23, #40).
Membranous β-catenin staining, representing the normal protein localization, was detected in 30 HCC samples (55.6%). The mutation T41I was found in one of these samples with membranous staining (#61). Finally, the β-catenin protein was not detected in a single HCC specimen (#14).
Regarding the correlation with biomarkers of infection, 10/12 (83%) of HCC samples positive for HBV presented a strong β-catenin accumulation (Score +++/++++) in nucleus or in cytoplasm. The phenomenon was significantly much weaker in HBV-negative tumors (2/8, 25%, p = 0.0161). Likewise 6/8 (75%) HCC samples with HCV biomarkers displayed nuclear or cytoplasmic β-catenin accumulation whereas a similar pattern was found in 54% of HCV (-) HCC samples (Table 2). This difference was not significant. Taken together, these data suggest that HBV is capable to trigger a nuclear or cytoplasmic accumulation of β-catenin even in absence of CTNNB1 mutation.
Distribution of β-catenin staining scores according to its subcellular localization in 54 HCC specimens from Colombia.
| CTNNB1 staining | P value | ||
|---|---|---|---|
| Weak (−/+/++) | Strong (+++/++++) | ||
| Cytoplasmic | |||
| Patients age | 69.0 ± 1.8 | 62.4 ± 1.6 | 0.01 |
| HBV biomarker | Nuclear and/or cytoplasmic | ||
| Positive | 2 (17) | 10 (83) | 0.016 |
| Negative | 6 (75) | 2 (25) | |
| Edmonson-Steiner Grade | Membranous | ||
| I-II | 22 (52) | 20 (48) | 0.05 |
| III-IV | 2 (17) | 10 (83) | |
We subsequently wanted to assess the correlation between tumor differentiation and β-catenin subcellular localization or staining intensity. Two groups G1-G2 and G3-G4 were considered. No subcellular delocalization was observed. By contrast, poorly differentiated tumors (G3-G4) were displaying more frequently an intense β-catenin staining at the membrane (mean score = 3.2 ± 0.2) than well-differentiated samples (mean = 2.4 ± 0.1, p = 0.033) (Table 2). The TP53 mutations (exon 5, 6, 8) described in cases HCC #1, #20 and #21 was correlated with accumulation in nucleus and/or cytoplasm. None of these samples presented CTNNB1 mutations.
In order to identify distinct subsets of HCC defined by a combination of multiple parameters, we proceed to an unsupervised hierarchical clustering analyzing of β-catenin localization and intensity on the series of 54 specimens. This analysis revealed the presence of a particular subset of HCC (n = 13, 24%) characterized by a systematic over the average cytoplasmic staining of β-catenin. This cluster was significantly associated with young age (77 vs. 32% of patients were below the median age, p < 0.0001 (Figure 3).

Class discovery analysis of the 54 HCC for the three subcellular localization compartments of beta-catenin using an unsupervised hierarchical clustering analysis. On the heatmap, blue colors correspond to below the average IHC intensity whereas reddish colors corresponds to above the average intensity of staining. Square above the heatmap correspond the different features. The yellow square on the dendrogram identifies the cluster defining a subclass of Colombian HCC with systematically high cytoplasmic beta-catenin staining and significant enrichment inyoung patients. Two out of the 3 CTNNB1 mutated samples are localized in this specific HCC subset. P values of Fischer exact tests corresponding to each enriched characters are provided below the heatmap.
The molecular mechanisms in liver carcinogenesis are not completely understood, mainly due to heterogeneity of etiology as well the heterogeneity in patients. However, several studies have confirmed that among the signaling cascades the Wnt/β-catenin pathway plays a key role in hepatocarcinogenesis. This pathway is critical in liver development, adult tissue homeostasis and regeneration by regulation of cell proliferation, growth, survival, oxidative stress and differentiation.
Aberrant activation of wnt signaling leads to expression of β-catenin/TCF dependent target genes that are critical for cell cycle, apoptosis and motility 4,18, 22,23
The modifications of the Wnt/β-catenin pathway described in HCC included genetic and epigenetic changes of CTNNB1, AXIN-1, AXIN-2, APC, CDH1, Frizzled, Secreted frizzled-related proteins (SFRP), Wnt and Dvl genes.4,17,24–30
In the current study, we have analyzed the Wnt/ β-catenin signaling pathway in 54 HCC cases from Colombian patients by characterization of mutations of CTNNB1 gene exon 3 and subcellular localization and expression of b-catenin protein.
We identified missense mutations in exon 3 of the CTNNB1 gene in 3/54 of HCC cases. Three mutations were identified at phosphorylation and kinases binding sites (P44S, S45P, T41I) and one additional mutation was detected in an ubiquitination-target motif of β-catenin (G34E). The mutation P44S was not previously reported in HCC cases.
These mutations interfere with phosphorylation of β-catenin protein by the complex Axin-APC-CK1 a-GSK3-β and subsequent proteasomal degradation. The genetic modifications of CTNNB1 gene imitate the activation of the Wnt signaling pathway and following stabilization of β-catenin protein in the cytoplasm. Accumulated β-catenin in the cytoplasm results in its translocation to the nucleus where this protein forms a complex with Tcf/Lef transcription factors and up-regulates the expression of target genes.31,32
The frequency of CTNNB1 (5.6%) in this series of HCC cases from Colombia, is the first one reported in a Latin American country and one of the lower frequency described over the world. This mutation frequency is similar to the results of previous studies of HCC cases in China and in Korea. In these countries as well in Malaysia and Taiwan, the CTNNB1 is ranging from 0 to 16% [Malaysia (0%), China (7.4%, 8%, 12%), Korea (2.8%, 16%) and Taiwan (13.1%)]. Additionally, these countries are in common that HBV infection is the main HCC risk factor.5,15,33–39
In Colombia, HBV infection was also the main risk factor in HCC (58.1%) according to the first study of epidemiological features of HCC.19,28,40
On the other side, the highest frequencies of CTNNB1 mutations have been reported in populations where the main HCC risk factors are HCV infection, alcohol and liver metabolic disease. The CTNNB1 mutation frequency is ranging from 19% to 40% in France (19%, 40%), Germany (25%), Japan (41.7%), Italy (17.5%) and USA (19.2%).14,17,24,41,42
Additionally, in one study of HCC cases from Japan, Switzerland and France, the frequency of CTNNB1 mutations was 31% in cases associated with HCV infection, compared with 19% and 13% associated with HBV infection or excess alcohol intake, respectively.43 Moreover, in a study of 22 HCC cases associated with HCV infection, CTNNB1 mutations were detected in 41% of tumors and in another study these mutations were exclusively detected in HCV-related HCC.44 These studies indicated that Wnt/β-catenin activation in a mutation-dependent way is related with the development of HCC in the context of HCV infection and non-viral HCC.44
Interestingly, Laurent-Puig, et al. evaluated the relation of genetic alterations, including CTNNB1 mutations, risk factors and survival in a series of 137 HCC. The results of this study provide evidence of two main pathways of liver carcinogenesis; the first group was defined by chromosomal instability, allelic losses (LOH) of 5 chromosome arms and mutations in Axin 1 and p53 genes mutations; additionally, this group was closely related to HBV infection and with poor differentiates tumors. In fact, HBV can induce chromosome instability and insertional mutagenesis.41
On the other side, non-HBV tumors were characterized by chromosome stability, 8p LOH and CTNNB1 mutation. The last two genetic alterations were described in most of the HCC cases without other LOH or mutations; this fact suggested that 8p LOH and CTNNB1 mutation could be early events in liver carcinogenesis.41
The experimental evidence of the role Wnt/β-catenin signaling activation in HCV-related HCC had been showed in different studies. The first evidence was the proliferation of Huh-7 human hepatoma cells induces by HCV Core protein. Indeed, this viral structural protein can modulate hepatocyte proliferation and transformation by regulation of signaling pathways, including Wnt/β-catenin pathway. Subsequently, the results of Liu, et al. confirmed the HCV Core effect on this pathway by three important findings; the accumulation of β-catenin through inactivation of GSK3-β, the enhancement of Tcf-dependent transcriptional activity induced by Wnt 3a and the in vivo evidence of tumor develop in athymic nude mice expressing HCV Core protein and Wnt-3a.45,46 Recently, Zhang, et al. demonstrated the critical role of MicroRNA (miRNA) 155 promoting hepatocyte proliferation and tumorigenesis via Wnt/β-catenin signaling activation. Indeed, the upregulation of miRNA-155 is a consequence of HCV infection and is dependent of nuclear factor kappa B.45,46 This interesting study offered a linkage between inflammation and tumorigenesis for the HCC associated with HCV infection. The chronic inflammation is a crucial event in the development of primary liver tumor in the context of HCV and/or HBV infection.
However, aberrant activation of Wnt/β-catenin signaling in hepatic tumorogenesis is due to not only CTNNB1 mutations but also Axin-1 and Axin-2 genes mutations, inactivation of GSK3-β, upregulation of Frizzled receptors and Wnt and methylation of Secreted Frizzled-related Protein (SFRP) gene promoters.41
Indeed, the protein HBx of HBV has been implicated in epigenetic modifications of SFRP1 and SFRP5 gene promoters. The silencing of Wnt antagonist genes in HCC and hepatoma cell lines was described first for Takagi, et al. SFRP promotor methylation was detected not only in HCC, but also in chronic hepatitis and cirrhosis cases suggesting that this mechanism is an early event of hepatocarcinogenesis.47 Recently, it was confirmed the down-regulation of SFRP1 and SFRP5 at the transcript and protein levels in HBV-HCC samples. Additionally, DNA methyltransferase 1 (DNMT1) overexpression in HBV-HCC cases was detected. The mechanism of this molecular event was demonstrated in vitro in HBx-expressing hepatoma cells; the viral protein HBx induced epigenetic repression of Wnt antagonists mainly attributed to recruitment of DNMT1 and DNMT3 to the SFRP1 and SFRP5 promoters producing constitutive activation of Wnt signaling pathway.48
Besides this epigenetic mechanism, additional evidence of the β-catenin up-regulation related with HBV infection are the stabilization of β-catenin protein by HBx-activated Src kinase probably by suppressing GSK3-β; and the protein-protein interaction between HBx and APC demonstrated in human hepatoma cells; the viral protein binds to the domain of APC necessary for its β-catenin association producing the displacement of β-catenin from the complex (Axin, APC, CK1α, GSK3-β) and disrupting its degradation.49
The Wnt/β-catenin pathway activation in HBV-HCC is in keeping with the results of the current study. Indeed, 83% of HBV-HCC samples showed a strong β-catenin accumulation in nucleus or in cytoplasm revealing activation of the Wnt pathway; this activation may be related with the properties of HBx protein as silencing of Wnt antagonist genes, protein-protein interaction APC-HBx and activation of Src kinase.
One of the variables that could be related to Wnt/ β-catenin pathway activation in HCC is the tumor differentiation grade. Some authors have suggested a correlation between CTNNB1 mutations, better cellular differentiation and favorable prognosis of HCC. However, the results of the different studies are contradictory probably due to different risk factors, genetic background of the patients and dissimilar histopathology and clinical criteria.35,39,49,50 In the present study, the grade of tumor differentiation of the three HCC cases with CTNNB1 mutations was GII (#23, #61) and GIII (#40).
Considering β-catenin localization, nuclear (N) and/or cytoplasmic (C) accumulation of this protein was detected in 42.6% of HCC cases (N 18.5%, C 24.1%) suggesting Wnt/β-catenin signaling activation. However, the tumor differentiation grade was similar of these HCC cases compared to tumor liver samples with membrane β-catenin localization (M). Interestingly, poorly differentiated tumors (G3-G4) displayed intense β-catenin staining at the membrane than well-differentiated samples. Unfortunately, the data of tumor size and survival of the patients was not available in order to analyze the correlation between these variables.
In one HCC sample (#14) the β-catenin protein was not detected as recently described for Lee, et al. in 13/89 HCC cases. These 13 cases showed significantly less inflammation and fibrosis. However the mechanism for β-catenin loss in HCC is unknown.50
The Wnt/β-catenin pathway activation in hepatic tumorigenesis is evidently one of the most important events in HCC. The pathway activation has been demonstrated in several studies not only by description on HCC samples but also in different in vitro models. Although, the interpretation and understanding the phenotype of β-catenin activation is quite difficult; indeed, it is necessary to take in account the heterogeneous mechanisms of β-catenin activation in HCC, the interaction with other signaling pathways, variability of HCC etiology and dietary factors. Additionally, there is not available data to answer the question if the target genes are the same regardless the mechanism of β-catenin activation and if the markers of pathway activation used in the different studies in clinical samples have the same weight.
In summary, this study describe for the first time the Wnt/β-catenin signaling activation in HCC cases from a Latin American country. Taking in account both the CTNNB1 exon 3 mutations and the subcellular localization and level expression of β-catenin protein, 44.4% of HCC samples obtained from Colombian patients showed Wnt/β-catenin signaling activation. In addition, Colombian patients represent the first population of HCC patients living in an inter-tropical region where Wnt/β-catenin pathway activation is quantitatively dominant over p53 alterations. This observation deserves further studies to generate additional biomarkers establishing the link between local risk factors and tumor phenotype.
AcknowledgementsWe thank Dr. Anne Lise Haenni from the Institut Jacques Monod for critical reading of the manuscript and Dr. Andres Arias from Universidad de Antioquia for discussion of the results. We also thank Ghyslaine Martel from International Agency for Research on Cancer (IARC) and Beatriz Vieco from Departamento de Patología, Universidad de Antioquia, for technical assistance.
Abbreviations- •
AFB: aflatoxin B.
- •
APC: adenomatous polyposis coli.
- •
C: cytoplasmic staining.
- •
CK1a: serine-threonine kinase-I alpha I.
- •
FSB: Fundacion Santafe de Bogota.
- •
Fz: Frizzled.
- •
G1: well differentiated.
- •
G2: moderately differentiated.
- •
G3: poorly differentiated.
- •
G4: undifferentiated.
- •
GSK3-β: glycogen synthase kinase 3β.
- •
HBV: hepatitis B Virus.
- •
HBx: HBV X protein,
- •
HCC: hepatocellular carcinoma.
- •
HCV: Hepatitis C Virus.
- •
HPTU: Hospital Pablo Tobon Uribe.
- •
LRP5/6: low density lipoprotein receptor-related protein 5/6.
- •
M: membranous staining.
- •
N: nuclear staining.
- •
Tcf/Lef: lymphoid enchancer factor/Tcell factor.
- •
UdeA: Facultad de Medicina, Universidad de Antioquia.
This study was funded by Departamento Nacional de Ciencia, Innovación y Tecnologia Colciencias (Grant 111540820471) and Proyecto de Sostenibilidad, Vicerrectoría de Investigación, Universidad de Antioquia.



