







Well-designed clinical trials are the gold standard for evidence-based research and for the assessment of the effectiveness of a clinical intervention. Methodological guidelines are available from various sources, such as textbook, funding applications and institutional guidelines. Nevertheless, a high number of published trials still lack methodological rigor, decreasing their utility, quality and scientific validity. In this article, we aim at providing some methodological recommendations for the development and report of a clinical trial, including the definition and recruitment of the sample, the basic study designs, randomization, blindness, data analysis and data report. Finally, we will discuss some of the most important ethical issues.
Los ensayos clínicos bien diseñados son el estándar por excelencia para la investigación basada en la evidencia y para la evaluación de la eficacia de una intervención clínica. Las directrices metodológicas se encuentran disponibles en varias fuentes, tales como libros de texto, solicitudes de financiamiento y directrices institucionales. Sin embargo, un gran número de ensayos publicados todavía carecen de rigor metodológico, disminuyendo su utilidad, calidad y validez científica. En este artículo, nuestro objetivo es proporcionar algunas recomendaciones metodológicas para el desarrollo e informe de un ensayo clínico, incluyendo la definición y selección de la muestra, los diseños básicos de estudio, la aleatorización, el cegamiento, el análisis y el reporte de datos. Finalmente, discutiremos algunas de las consideraciones éticas más importantes.
Clinical trials (CTs) are prospective studies that aims at investigating the effects and value of a new intervention on a specific population, over a defined period. The intervention can be of different nature (like medical, pharmacological or behavioral) and can have either preventive, therapeutic or diagnostic purposes.
When adequately designed, performed and reported, CTs are the gold standard for evidence-based research (Moher, Schulz, Altman, & Group, 2001). For these reasons, all trials should meet some important methodological criteria (Moher et al., 2001): Lack of procedural rigor may lead to biased results, which are difficult to consider valid, generalizable and reliable (Juni, Altman, & Egger, 2001).
Every year, dozens of CTs are published; by the way, up to 50% of them show important methodological deficiencies (Chan & Altman, 2005). The same negative trend has been observed in the psychological field (Michie et al., 2011; Stinson, McGrath, & Yamada, 2003). Aiming at improving the quality of reports, an international group of clinicians, statisticians, epidemiologists and biomedical editors have created the CONSORT (CONsolidated Standards Of Reporting Trials) statement, which consists of a checklist and a flow diagram for reporting CTs (Begg et al., 1996). Subsequently to the original version, some revisions have been published: The last revision dates back to 2010 (Schulz, Altman, Moher, & Group, 2010), Other than providing a methodological systematization, the CONSORT statement constitutes a valid tool that allows readers to be able to evaluate by their own the quality of a CT. Nevertheless, many behavioral investigators have not completely adopted these guidelines (Bonell, Oakley, Hargreaves, Strange, & Rees, 2006; Stinson et al., 2003), considering them not fully adequate for the investigation of social and psychological interventions (Mayo-Wilson, 2007). For instance, explicit guidelines related to external validity and process evaluations are still missing (Armstrong et al., 2008; Prescott et al., 1999). For these reasons, more specific guidelines have been created: An attempt in this direction are JARS (Journal Article Reporting Standards), developed by the American Psychological Association (APA) (Publications & Communications Board Working Group on Journal Article Reporting, 2008), or the CONsolidated Standard Of Reporting Trials – Social and Psychological Interventions (CONSORT-SPI), which is being developed by the Centre for Evidence Based Intervention at the University of Oxford, the Centre for Outcomes Research and Effectiveness at University College London, and the Institute of Child Care Research at Queen's University Belfast, in association with the CONSORT Group (Montgomery et al., 2013).
Every well-designed CT requires a protocol, a written agreement where the key points of the study are exposed. Importantly, all protocols should be defined before the beginning of the trial and they should be not modified anymore in the subsequent phases.
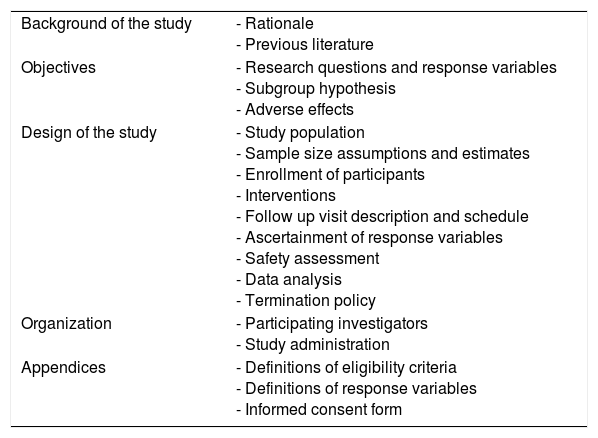
The main topics that every protocol should address are listed in Table 1 (Friedman, Furberg, DeMets, Reboussin, & Granger, 1998). Starting from this scheme, the main phases of the development, application and results’ reporting of a CT will be discussed in the next paragraphs. The reported methodological recommendations are based on a synthesis of the existing guidelines identified in literature.
Schematic key points of a clinical protocol.
| Background of the study | - Rationale - Previous literature |
| Objectives | - Research questions and response variables - Subgroup hypothesis - Adverse effects |
| Design of the study | - Study population - Sample size assumptions and estimates - Enrollment of participants - Interventions - Follow up visit description and schedule - Ascertainment of response variables - Safety assessment - Data analysis - Termination policy |
| Organization | - Participating investigators - Study administration |
| Appendices | - Definitions of eligibility criteria - Definitions of response variables - Informed consent form |
First of all, it is important to be clear and explicit about the rational of the study and to ensure that there is consistency between the theoretical stance and the developed protocol (Twining, Heller, Nussbaum, & Tsai, 2016). The definition of the theoretical background includes the analysis of literature and the explanation of the scientific background, in order to examine and compare what investigators have already pointed out on the same topic.
In this early planning phase, investigators should define what the experimental study wants to give an answer to, namely defining the research questions.
Research questions are the fundamental core of every research study and they should be selected and defined in advance, being as specific as possible. The primary question is the most important issue the study wants to answer to and it is typically a test of the effect of a specific intervention (Ellimoottil, Vijan, & Flanigan, 2015): The entire CT is then developed based on it. On the other hand, secondary questions are subordinate questions and they are usually related to the primary question. They can be differentiated in two categories: (1) secondary questions in which the response variable is different than the one of the primary question and (2) secondary questions that are related to subgroup hypotheses. Finally, investigators may also define some ancillary questions that, even if not directly related to the implemented intervention, could be addressed by the outcomes of the trial.
A response variable is the variable from which the researchers are expecting a specific answer. Response variable is then the variable that will be collected during the trial in order to respond to the research questions. They should be capable of unbiased assessment, be assessable in all participants and be ascertained as completely as possible (Friedman et al., 1998).
The response variable is also known as dependent variable: Indeed, the response variable depends on another factor, known as independent variable, whose variation is supposed to be the cause of changes in the dependent one. Obviously, the primary question may be best answered by more than just one response variable, i.e. “composite” or multiple response variables. In this case, investigators are able to determine if a specific independent factor has an impact on various other concepts.
When the endpoint of interest is too difficult and/or too expensive to evaluate, investigators may choose to use surrogate response variables. According to Prentice, three important rules should be followed so as to avoid possible biases (De Gruttola et al., 2001): (1) the surrogate variables must correlate with the true clinical outcome, (2) they must capture the full effect of the intervention, and (3) they should be accurately and reliably assessed (Prentice, 1989).
Regardless to their nature, appropriate endpoint should always be defined a priori and assessed in all patients by blinded reviewers (Friedman et al., 1998).
Definition of the samplePopulation study and inclusion criteriaThe definition of the specific targeted population (i.e. a subset of the general population characterized by specific features or conditions of interest) is one of the most important phases for the definition and interpretation of a CT, with significant repercussions on the selection of the inclusion and eligibility criteria. Indeed, one of the main purposes of this process is making the results generalizable (i.e. how far outcomes are representative of the entire target population). The population study and the definition of inclusion and exclusion criteria should precede the definition of the protocol and should always be clearly reported and justified (Friedman et al., 1998). Despite being so important, many studies do not always report the specific characterization of the sample in an adequate way (Van Spall, Toren, Kiss, & Fowler, 2007).
Among all, the investigators should always specifically and transparently declare the characteristics of the required participants: All the eligibility criteria should be clearly specified and defined considering five different dimensions (Table 2) (Friedman et al., 1998). If it is not possible, at least the most important criteria should be evidently exposed.
Dimensions to consider during the definition of inclusion/exclusion criteria.
| Benefits | Selection of patients who can surely benefit from the treatment. |
| Homogeneity | Considering criteria that allow for homogeneous samples. |
| Success | Considering criteria that allow for higher intervention success rate. |
| Risk and disadvantages | Deleting or at least minimizing possible risks, adverse factors or disadvantages for patients. |
| Adherence | Considering the voluntary adherence of participants in following the protocol. |
During the definition of the sample, investigators may consider the possibility of applying a stratification of the population, usually based on two or more prognostic factors which are supposed to make the groups imbalanced. Stratification and subgrouping may help investigators better identifying the underlying mechanisms of action of the considered intervention. Importantly, all subgroups analyses should be defined a priori.
Sample size and sensitivity analysisAnother fundamental point during the population study is the definition of the sample size, i.e. how many participants should be involved into the CT (Nirmalan & Thomas, 2007). Every trial should have sufficient statistical power to detect differences between groups. Therefore, the calculation of the sample size with prevision for adequate levels of significance and power is an essential part of the trial planning (Friedman et al., 1998). Sample size estimation should be determined a priori and it should always be transparently reported (Ellimoottil et al., 2015). However, most of the published CTs do not always contain sufficient information to allow other investigators to reproduce the sample size (Clark, Davies, & Mansmann, 2014).
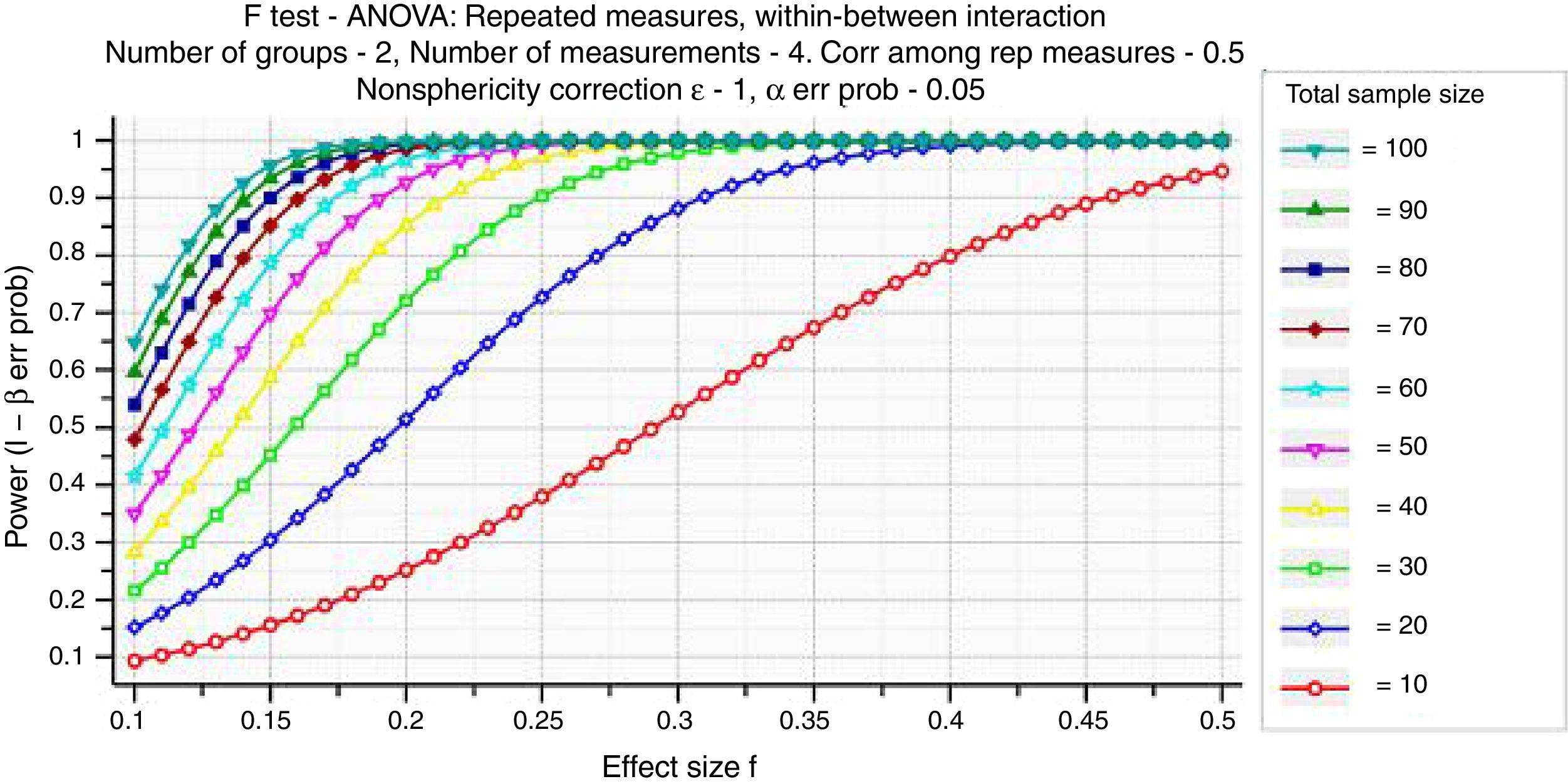
Hypothesis test tells us the probability of a result of that magnitude occurring, if the null hypothesis is correct (i.e. there is no effect in the population). It does not tell us the probability of that result, if the null hypothesis is false (i.e. there actually is an effect in the population).
Specifically, we consider the effect size, the sample size, and the criterion required for significance (α, where α is probability of Type I error). These three factors, together with power (1−β, where β is probability of Type II error), form a closed system – once any three are established then the fourth is completely determined (Fig. 1) (Faul, Erdfelder, Lang, & Buchner, 2007).

Investigators should always adopt a conservatory approach, considering the possibility of errors in the initial sample size estimation.
Recruitment of participantsSuccessful recruitment requires a specific plan with multiple strategies, aiming at obtaining the needed sample in a predetermined limited period. Nevertheless, investigators usually have to deal with many recruitment problems. According to Friedman, there are both scientific and logistic difficulties (Friedman et al., 1998):
- 1.
From a scientific point of view, recruitment of participants should be conducted into an optimal time window. Indeed, changes influencing the study may occur: The development of new interventions, changes in the targeted population or variation in the definition of the interested condition.
- 2.
From a logistic point of view, when recruitment is difficult and too prolonged, negative repercussions may impact on the study: Higher pressure, costs, frustration and discouragement.
Other observed problems are for instance related to erroneous sample estimations, such as overestimation of the possible target population or underestimation of needed time and efforts. More, participants may not be interested in entering a CT for many reasons, like inconvenience, concerns about the experimentation, perceived lack of benefits or simply fear (Barnes, 2007). In order to make this process easier, the investigators may choose to increase the involved geographical area, include other hospital or institutions in the recruitment of participants or even to revise some criteria. Pilot study can also be used to estimate the effect size of the treatment and draft a more precise sample size computation. Importantly, a widely known risk is the “healthy volunteer effect”: Individuals who are voluntary joining the trial may be different from non-participants and may therefore bias results.
The recruitment of subjects usually follows predetermined sampling schemes, which are selected depending on the research question. Among the most important schemes, investigators may adopt convenience sampling (recruitment of patients who are easier to access), simple random sampling (subjects are randomly selected from the entire population), stratified random sampling (random samples are selected after the determination of subgroups of interest) or cluster sampling (selection of subjects within a specific cluster).
One of the greater challenges in the recruitment process is the management of disease comorbidity: Generally, most of the individuals with a chronic disease show a coexisting medical condition. Notably, comorbidity may have a deep impact on the course of a treatment for different reasons: (1) greater difficulties in self-care, like coordinating different medications, or high economic costs; (2) influence on the frequency and intensity of treatments for coexisting conditions; (3) reduced adherence, compliance and tolerability for medication; (4) increased risk of drug-to-drug interactions (Bayliss, Steiner, Fernald, Crane, & Main, 2003; Redelmeier, Tan, & Booth, 1998). Accordingly, researchers tend to exclude patients with comorbidities from clinical trials. Even if this allows to obtain a more homogeneous sample, the obtained results may not be easily generalizable to the real population in terms of safety, tolerability and efficacy of the investigated treatment. Accordingly, RCTs may be classified in two categories. On the one hand, explanatory trials investigate the efficacy of a treatment by selecting participants under ideal conditions, and obtaining therefore an experimental sample as more homogeneous as possible (Thorpe et al., 2009). On the other hand, pragmatic trials aim at analysing the effects of a new intervention taking into consideration the high heterogeneity of real-world clinical populations. In that sense, these two types of design should not be considered as completely independent, but rather as the poles of a continuum. Accordingly, the Pragmatic-Explanatory Continuum Index Summary (PRECIS) (Thorpe et al., 2009) was created so as to assist researchers in defining all the factors that may influence the position of the trial within this continuum, including eligibility criteria and inclusion/exclusion of comorbidity.
The inclusion or exclusion of participants with a comorbidity poses therefore a great challenge, and require careful consideration by researchers and clinicians conducting a clinical trial. Their exclusion, indeed, could raise ethical concerns, whether their inclusion could influence results in terms of effectiveness, adverse effects and outcomes interpretation (Marrie et al., 2016). When patients with comorbidity are included, a possible solution is represented by the calculation of a comorbidity index, that represents in a single numeric score the co-existence and severity of other illness. By calculating this score for every single patient, researchers may provide clearer explanations on the effect of comorbidity on treatment outcome. One example in the Charlson Comorbidity Index based on the ICD-9 diagnosis codes (Charlson, Pompei, Ales, & MacKenzie, 1987). Alternatively, clinical trials may adopt performance indices, i.e. calculate the impact of a disease on the general health of a patient (Hall, 2006).
The inclusion of patients with comorbidities should not preceded by preliminary assessments (Marrie et al., 2016): Whether stricter criteria are required in the first trials phases to provide important insights on the safety, side effects and efficacy of a new treatment, phase II clinical trials should already start investigating the interaction of the intervention with possible comorbidities.
Study designPreliminary phasesDepending on the nature of the considered intervention, every CT is typically preceded by four preliminary phases. While pharmacological CTs usually require all the four preliminary phases, trials adopting a different intervention, such as a behavioral one, not always follow all these phases (Table 3).
Preliminary phases in CTs.
| Phase I studies | Estimation of tolerability (“maximum tolerated dose”) and safety of the intervention on a small sample. |
| Phase II studies | Evaluation of the biological activity, adverse effects and safety of the biomedical or behavioral intervention on a larger sample. |
| Phase III studies | Evaluation of the effectiveness, efficacy and adverse effects of the new intervention on a bigger sample compared to another experimental condition. |
| Phase IV studies | Evaluation of long-term effects of the new intervention on the general population; collection of information about adverse effects associated with widespread use. |
CTs may adopt different types of experimental designs. The most important basic study designs are reported in Table 4.
Basic study designs in CTs.
| Parallel design | The experimental and the control group are simultaneously followed. |
| Historical control studies | The experimental group is compared to a group who was previously administered a standard or control therapy. |
| Cross over trials | Participants take part at least once for each group, as they serve as their own control. |
| Withdrawal study | During the starting phase participants are assigned to the active intervention. Then, they are divided in two groups: A portion is followed on the active intervention, while the other is followed off the intervention. |
| Factorial design trials | Evaluation of more than one interventions, which are compared to a control condition. |
Every CT should compare the considered intervention to a control group, which can be for instance represented by a placebo condition or by the administration of another standard intervention. Importantly, the selection of placebo as control condition should always be clearly justified by investigators, explaining the reason why the standard intervention has not been compared.
The selection of the control condition depends on the main purpose of the study: While superiority trials aim at showing the greater efficacy of the new intervention, non-inferiority trials aim at demonstrating that the new intervention is not worse than the standard one. This latter design is usually preferred when the side effects of the selected intervention are supposed to be inferior to those of the standard intervention.
RandomizationRandomization is the process by means of which to assign participants to one of the experimental groups with a same level of chance (i.e. participants are equally like to be assigned to either the intervention group or the control group).
There are three main common randomization methods: (1) simple randomization, when participants are randomly assigned to groups, for instance based on coin flip or random number allocation; (2) blocked randomization, when participants are assigned to groups and then equally randomized within the groups, and (3) stratified randomization, when investigators stratify the sample and then randomize participants within the strata.
Randomization may follow two different rules. Through fixed allocation randomization, allocation probability in not altered throughout the study. Differently, adaptive randomization procedures allow changes in the allocation probability as the study progresses. Randomization should always be reported in the title of the trial (Schulz et al., 2010).
On the other hand, there is also the possibility to assign participants without using randomized processes (non-randomized CT).
According to many investigators, randomization is the gold standard to achieve scientific comparability between groups (Armitage, 1982). Anyway, both approaches show advantages and disadvantages (see Table 5). For these reasons, the applied randomization approach should always be clearly justified.
Some advantages and disadvantages of randomized and non-randomized CTs.
| Advantages | Disadvantages | |
|---|---|---|
| Randomized CT | (1) Produce comparable and balanced groups with respect to known or unknown risk factors. (2) Delete possible biases induced by investigators. (3) Validity of statistical tests of significance (ascribe a probability distribution to the difference in outcome between treatment groups receiving equally effective treatments and thus assign significance levels to observed differences (Byar et al., 1976). (4) Guarantee that statistical tests will have valid false positive error rate. | (1) Ethical reasons (Ingelfinger, 1972): Randomization is not possible when depriving a patient from a treatment that is believed to improve his condition. (2) Not possible when the prevalence of the targeted population is low. |
| Non-randomized CT | (1) Participants are not assigned depending on the chance. (2) Easier to create groups by matching characteristics. | (1) Groups may not be comparable. (2) Assignment may be influenced by the investigators. |
Possible errors in the randomization process may have negative repercussions on the entire study. Briefly, two different biases are possible: (1) selection bias, when the allocation process is predictable and consequently influenced by the anticipated treatment assignment, and (2) accidental bias, when the randomization procedure does not achieve balance on risk and prognostic factors.
BlindnessBlindness refers to whether information about the administrated intervention is known or unknown to both investigators and/or participants. Different types of blindness may be adopted.
In unblinded trials, both the participants and the researchers know the assignment to groups and therefore what type of intervention is being administrated. Differently, in single blind trials only the investigator knows the assignment of participants. Even if these approaches are very easy to adopt, both of them have a high risk of bias, as the investigators may consciously or unconsciously affect data. For this reason, the use of double blindness is usually preferred as neither the participants and the investigators know the assignment. Nevertheless, double blindness gives no protection against imbalanced groups and it loses its validity if participants or investigators discover the group assignment. Finally, a fourth option is the use of triple blindness, in which the committee monitoring response variables is not told the identity of the groups; the committee is simply given data for groups A and B.
Despite being important, information about blindness is not always adequately reported (Schulz, Chalmers, Hayes, & Altman, 1995). According to the CONSORT statement, two information about blindness should be reported in every CT: “if done, who was blinded after assignment for interventions and how? If relevant, description of the similarity of interventions” (Schulz et al., 2010). Among the most important advantages, providing precise indications about blindness gives to the investigators and to the readers the idea of how much confidence should be given to the observed results.
Data collectionBaseline assessmentTo evaluate the value and the effects of an intervention, participants should always be assessed at baseline (i.e. the status of subjects at the beginning of the trial). Baseline may be assessed through different tools depending on the research questions and on the nature of the intervention, such as interviews, questionnaires, physical examinations or laboratory tests. Some variables should be always considered into the baseline, regardless to the intervention, such as demographics, socioeconomics, risks or prognostic factors, medication load or medical history. Notably, it is always difficult to enroll newly diagnosed patients: One of the variable to be taken into consideration regards therefore medication and previous/current treatments. When interested in investigating a new drug, participants may be asked to be off all other medications. If during the baseline assessment this requirement is not satisfied, researchers may decide to repeat the procedure and ask participants to return for the baseline assessment in a week.
Without a baseline, it would not be possible to understand the real effects of the intervention, to generalize results or even to compare the CT results to other trials which adopt the same intervention. Moreover, baseline assessment is important to comprehend whether the groups are comparable before the intervention, to find out which variables may influence the outcomes, and to determine whether groups are unbalanced (and if they do, whether a particular trend of imbalance may influence results) (Friedman et al., 1998).
One of the most critical points is to decide when to assess the baseline. Generally, the time between recruitment and baseline evaluation should be as shorter as possible, in order to avoid possible changes in participants’ status: Indeed, it is not possible to control for adverse events that may occur between these two phases. These uncontrollable variables may influence baseline measurements and decrease the power of the results. When changes at baseline occur before the randomization, participants should be excluded from the study. Otherwise, participants should be kept in the trial, taking obviously in consideration it during the analysis.
Data analysisData analysis is usually underestimated, and considered as a fast and effortless process. Nevertheless, this is probably one of the most critical phases of the entire RCT: Good statistical knowledge is crucial in creating a clinical trial, and in selecting the correct statistical tests. Incorrect choices during this step could indeed introduce important biases, and result in misleading conclusions.
The “intention to treat” (ITT) is one of the most important principle of a CT, according to which after randomization patients should be analyzed based on their initial assigned group, and not on the actually treatment received. This definition is counterposed to “per-protocol” and “as-treated” designs, where in the final analysis are included just the patients who completed the protocol they were initially assigned to.
Every CTs use successive observations of the same variable (measure considered for the analysis) on each subject. Repeated measures are defined as measurements sequentially conducted in time (temporal factor) or location (spatial factor) on the same subject. Repeated measurements are commonly employed to estimate measure parameters, investigate the factor effect on the process, and model and monitor the production and its process (Lee & Gilmore, 2006). It is highly suggested to follow the recommendation of Bakker and Wicherts in reporting statistical results (Bakker & Wicherts, 2011).
Among the main methodological problems that many CTs have to deal with, there are short-term follow up and limited number of comparison groups: In these situations, simulation models may be adopted in order to increase the interpretation of the outcomes (Caro, Briggs, Siebert, Kuntz, & Force, 2012). Nevertheless, more longitudinal RCTs should be performed. According to the World Health Organization (WHO (World Health Organization), 2011), longitudinal studies play a fundamental role for the understanding of the “dynamics of disability”, especially when considering studies in the field of rehabilitative interventions. However, a recent systematic review pointed out that, taking into account RCTs in post-stroke rehabilitation, only 39% of the studies performed longitudinal analysis of data, whether the 52% ignored the longitudinal nature of data (Sauzet, Kleine, Menzel-Begemann, & Exner, 2015). Moreover, 95% of RCTs published in top medical journals show different levels of missing data (Ashbeck & Bell, 2016). Whether attrition of less than 5% is unlikely to introduce bias, higher levels of attrition may bias the validity of a study (Sackett, 2000). In that sense, the longitudinal nature of data should always be considered into repeated measures analysis and the intervention effect over the follow-up should always be reported. This could be achieved by mean of statistical methods like mixed models (Sauzet et al., 2015). In this perspective, also epidemiological studies and related time series analyses, using temporal and spectral methods, should be considered as a keen instrument to be included as a toolkit in clinical sciences. Last but not least, the use of Bayesian methods should be increased also for clinical trials, due to well-known problems of the p-value misuses (Baker, 2016).
Power analysisOnce the results are computed a power analysis can be used to anticipate the likelihood that the study yielded significant effects. In particular, the goal of a post hoc power analysis is to compute achieved power, given the effective other three factors (sample size, achieved power and significance level), which can be read or deducted by data (output of statistical data analysis). Since many statistical software give pη2 (partial eta-square) values instead of Cohen's f effect size, it is important to compute f=sqrt[η2/(1−η2)], where sqrt is for square root calculation.
According to post hoc power analysis, some significance level could be high informative even if slightly higher than 0.05 (it depends on achieved pη2 for that measure).
Data reportThe final phase of every CT is the report and interpretation of the obtained outcomes. Authors are in a privileged position, as they exactly know all the strengthens and weakness of their study: For this reason, they have the responsibility to critically report and review results, to avoid erroneous interpretations and to declare possible limitations (Schulz et al., 2010). Moreover, authors should always declare all real, potential or apparent conflicts of interest. Regrettably, up to 50% of published trials show erroneous reporting, including deficiencies in reporting randomization, sample size estimation and primary endpoints definition (Chan & Altman, 2005).
Among the main responsibilities, investigators should always report all results, whether positive, neutral or negative, as also efficacy and safety parameters. Unfortunately, biases in many publications are often observed: In the psychological field, up to 97% of published articles show positive results, i.e. a confirmation of the starting hypothesis (Sterling, 1959). Indeed, the problem of selective reporting is one of the most critical issue in CTs (Al-Marzouki, Roberts, Marshall, & Evans, 2005), mainly due to the fact that journals are more likely to publish positive rather than negative or neutral outcomes (Pocock, 2013). According to Friedman, there should be on one side a greater responsibility by authors in reporting all results, regardless to their direction; on the other side, journals should encourage full and honest reporting (Friedman et al., 1998).
As already noted above, information about blindness, randomization, adherence and concomitant treatment should always be precisely reported. Primary endpoints should be reported before secondary endpoints. In addition, exploratory endpoints and post hoc analysis should not be addressed before the report of primary and secondary endpoints.
As previously underlined, CTs are the core of evidenced-based research. Consistently, investigators should provide this information in order to make readers able to judge by themselves the validity of the reported outcomes: When adequately conducted, a CT can provide clinical evidence that could modify and improve the current clinical practice. A clear data report is also fundamental in order to place results into the existing scientific background, to open new possible future research directions and to comprehend whether results are possible to generalize to the entire population.
Ethical issuesEthical aspects of CTs has been deeply debated. Investigators and sponsors have indeed ethical obligations to participants, as to science and medicine (Friedman et al., 1998). During the development of a protocol, seven different ethical dimensions should always be considered (Emanuel, Wendler, & Grady, 2000) (Table 6).
Ethical issues in a CT (Emanuel et al., 2000).
| Value | Evaluation of the scientific and social value of the considered intervention. |
| Scientific validity | Adherence to methodological rules, to obtain valid and reliable results. |
| Fair selection of participants | Clear process of selection and recruitment, with consideration of vulnerable patients and of benefits for sponsors. |
| Favorable benefits and risk balance | Minimization of risks and adverse effects, along with maximization of benefits. |
| Independent review | Review of the protocol by unaffiliated investigators. |
| Inform consent | Clear information for participants before joining the trial. |
| Respect for participants | Permitting withdrawal, protecting privacy, informing participants of possible new risks and of clinical results, keeping welfare of participants. |
During the definition of the research question, investigators should evaluate whether the research questions are enough important to justify possible benefits, adverse effects or even possible risks. Importantly, recruitment should obviously consider participants health: Randomization, for instance, is not adoptable when other better interventions exists. The same observation should be considered when defining the control group. Among all, placebo is one of the most debated decision (Freedman, Weijer, & Glass, 1996). In the placebo condition, participants are usually administrated with substances or interventions that are supposed not to be effective. Accordingly, this condition is particularly used to better control for the effects of a specific treatment, more than guaranteeing for blindness. However, the ethical discussion around the adoption of placebo is about ten years old (Spławiński & Kuźniar, 2004). On the one hand, the use of placebo in superiority trials allows to unequivocally ensure the effects of a new treatment. On the other hand, placebo seems to violate the uncertainty principle of the informed consent and to be unethical, as patients would be prevented from receiving an already available efficacious intervention (Rothman & Michels, 1994). The use of placebo poses another methodological issue in results interpretation: The new intervention could be better than the placebo condition, but not better than the gold-standard treatment. To clarify the choice of a specific comparator when conducting a clinical trial, the last version of the Declaration of Helsinki states that “The benefits, risks, burdens and effectiveness of a new intervention must be tested against those of the best proven intervention(s), except in the following circumstances: Where no proven intervention exists, the use of placebo, or no intervention, is acceptable; or Where for compelling and scientifically sound methodological reasons the use of any intervention less effective than the best proven one, the use of placebo, or no intervention is necessary to determine the efficacy or safety of an intervention and the patients who receive any intervention less effective than the best proven one, placebo, or no intervention will not be subject to additional risks of serious or irreversible harm as a result of not receiving the best proven intervention. Extreme care must be taken to avoid abuse of this option” (World Medical Association, 2013). Even if “permissible” in some particular conditions (Millum & Grady, 2013), the use of placebo still remains controversial, and the adopting of active-control studies has been proposed as a more ethical solution (Spławiński & Kuźniar, 2004).
The informed consent is a fundamental but not a sufficient tool in order to guarantee the respect of participants. Through the consent, investigators clearly expose all the key points of the trial, along with possible implications, risks or benefits. It should also declare whether data will be shared with other researchers or will be used also for other purposes. Investigators should update and inform patients whether safety, risks or benefits change during the trial: If safety decrease, the trial should be stopped.
Many types of informed consent have been developed, like the Nuremberg Code, the Declaration of Helsinki or the Belmont Report. In special situations, such as the recruitment of minors or emergency situations (for example, when patients are not fully able to comprehend the informed consent), specific guidelines that has to be followed.
ConclusionCTs are the fundament of clinical practice: They can provide important evidence in order to change the current state of art and ameliorate patient care. To increase the transparency and value of publications, different guidelines have been developed; the CONSORT statement, for instance, has considerably improved report quality (Lucy Turner et al., 2011). Moreover, guidelines are essential for readers to evaluate and make best use of new evidence (Montgomery et al., 2013).
Guidelines for CTs are available from different sources, such as textbook, funding applications and institutional guidelines (Tetzlaff et al., 2012). Nevertheless, a high number of published CTs still lack of methodological details, decreasing their utility and scientific validity (Tetzlaff et al., 2012). This mainly occurs in the psychological field, where CTs are usually poorly reported. Aiming at improving mental health, social and psychological interventions are very complex, as characterized by multiple components influencing the clinical outcome at different levels (Ribordy, Jabes, Banta Lavenex, & Lavenex, 2013). These characteristics make the current guidelines, including the CONSORT statement, not fully appropriate for psychological interventions. Consequently, the CONSORT-SPI statement represents an important step toward the definition of more specific guidelines for reporting trials in psychological research (Grant, Mayo-Wilson, Melendez-Torres, & Montgomery, 2013).
Conflict of interestThe authors declare that no competing interests exist.
This work was partially supported by the Italian funded project “High-end and Low-End Virtual Reality Systems for the Rehabilitation of Frailty in the Elderly” (PE-2013-02355948).
