




Introduction
Recent evidence suggests that agonists of opioid receptors induce cardioprotection.1,2 Cardioprotection can be produced pharmacologically through different pathways. To this regard, Schultz et al. (1995) demonstrated that opioids produce cardioprotection through a G-protein-linked mechanism in a myocardial infarct rat model.3 Miki et al. demonstrated that the cardioprotection induced by morphine in the myocardium of rabbits is mediated by the protein kinase C (PKC).4 Opioids can induce a mechanism mediated by multiple kinases similar to those involved in the preconditioning of ischemia. Among the reported kinases are the PKC, the tyrosine kinase, and a family of mitogen-activated protein kinases (MAP kinases).5 Regarding the final effect of cardioprotection, involvement of ATP-modulated potassium channels in the cellular membrane (KATP) and in the mitochondria (mito KATP) is known. It has been demonstrated that TAN-67 an agonist of the delta-1 receptor produces a potent antiarrhythmic and antifibrillatory effect on ischemia that can be abolished by KATP antagonists in the mitochondria.6 KATP inhibitors, such as glibenclamide, can abolish this cardioprotection.7 Agonists of opioid receptors can also have antiarrhythmic properties as part of their cardioprotective effect. Márquez Montes et al. demonstrated a beneficial effect of morphine on the electrical fibrillation threshold of the ventricle in anesthetized dogs;8 these authors used morphine (3 mg/kg IV) and elevated the fibrillation threshold from 10 mA to 20 mA. Fentanyl, an agonist of the mu-receptor, increases the ventricular fibrillation threshold in 14% in anesthetized dogs.9,10 Yu et al, in 1999, suggested that the stimulation of kappa-opioid receptors produces cardioprotection against arrhythmias and intracellular calcium oscillations induced by beta-adrenergic stimulation.11 Nekrassov and Peón, from the Instituto Nacional de Cardiología “Ignacio Chávez”, studied the effect of conorphone (3 mg/kg IV) on an experimental myocardial infarction in rats by administering the drug 10 min before ligating the anterior descending coronary and found a statistically significant decrease in the infarct size. Besides, by electron microscopy, these authors observed intracellular edema and destruction of crests and membranes of the mitochondria, in the control group. However in the conorphone-treated group they observed mitochondrial vacuoles without other manifestations of cellular damage.12 Recently, Cho et al. demonstrated that opioid peptides that act on the mitochondria, diminish the myocardial infarction in rats induced by ischemia-reperfusion, and arrhythmias were less frequent and severe than in the control group.13
Remifentanil is an opioid of the 4-anilido-piperidine family with an agonist effect on mu, delta, and kappa receptors.14 Remifentanil presents a short elimination half-life of approximately 3min to 10 min; its chemical structure has an ester linkage that undergoes rapid hydrolysis by non-specific tissue and plasma esterases.14 It is widely used in surgery for its pharmacokinetic properties and because the recovery from remifentanil anesthesia is much faster than with any other opioid studied until now.15
The objective of this study was to determine if remifentanil exerts antiarrhythmic action in experimental arrhythmias in the dog (atrial flutter, atrial ectopic focus induced by aconitine and in arrhythmias induced by digitalis intoxication) and to assess its cardioprotective effect by measuring the time course of a lethal dose (LD) of digoxin.
Material and methods
We studied 25 mongrel dogs of either sex, weight between 12 kg and 18 kg, anesthetized with sodium pentobarbital at a dose of 30 mg/kg applied IV. Tracheotomy was performed for pulmonary ventilation with 30 ml/kg of ambient air (Palmer CF ventilator, Mod. London 8W2). Arterial blood samples were taken to measure pH, PCO2, and PO2 before, during, and at the end of the experiment to corroborate that pulmonary ventilation was adequate. The chest was opened through median sternotomy and the pericardium was dissected to expose the heart and place four pairs of recording and stimulating electrodes: two pairs in the right atrium and two pairs in the right atrial appendage (RAA). Four more electrodes were placed in the limbs for the electrocardiographic recording of lead DII. To record arterial pressure the right carotid artery was dissected and cannulated and connected to a U-shaped Hg manometer. A Swan-Ganz catheter was passed through the femoral artery to the left ventricle to record the intraventricular pressure (IVP). The femoral vein was cannulated for the administration of fluids (Hartman solution, 100 ml/h), anesthesia, and the drugs to be studied. A VR6 Electronics for Medicine polygraph with interphase for data acquisition in a personal computer was used.
Animals were divided in five groups. Group I (n = 5) received remifentanil at 0.1 and 1 μg/kg, to determine the dose-effect relationship on arterial pressure IVP, and heart rate (HR), as well as to find the dose with the minimal hemodynamic effect to be used in the other groups.
Group II (n = 5) received digoxin by means of an infusion pump at a speed of 9 μ/kg/min to determine the temporal course of digitalis intoxication and measure the time (min) to reach the LD.16
Group III (n = 5) received digoxin infusion (9 mg/kg/min) until reaching the first signs of digitalis intoxication (sustained A-V block, ventricular extrasystoles), after that remifentanil was administered as a single bolus. In two additional experiments, two remifentanil boluses were administered. The first bolus was given at the beginning of the digitalis intoxication, and the second one, 5 min thereafter.
Group IV (n = 5) received digoxin (9 mg/kg/min) to induce digitalis intoxication, once the early electrocardiographic intoxication signs have appeared, naloxone was administered at a single dose of 0.01 mg/kg. Afterwards, remifentanil was administered in a single bolus.
In group V (n = 5), we studied the effect of remifentanil in the double arrhythmia model. This method consists in inducing simultaneously two arrhythmias in the right atrium of the anesthetized dog with the open chest. By clamping the base of the atrial appendage, the rest of the atrium becomes electrically isolated. A very small aconitine crystal was placed near to the tip of the atrial appendage, which induces an ectopic focus with a frequency usually higher than 5 beats per second. In the remnant atrium, a flutter was produced by the circular movement of a stimulus following the method of Rosenblueth and García Ramos.17 This method is based on high-frequency electrical stimulation (20 Hz).18 These approaches yielded an experimental model of two arrhythmias of different mechanisms that were recorded in the same atrium. Figure 1: Once both arrhythmias have been produced, they were observed for 20 min to confirm their stability and to administer remifentanil in a single bolus. Results are presented as mean ± standard deviation (SD).

Figure 1. Double arrhythmia preparation diagram. Note the isolation zone of the right atrial appendage (RAA) (starting at the dotted line), the production of injury zone that prolongs the natural obstacle in the inferior vena cava and placement of electrodes. LA: left atrium; RA: right atrium; SVC: superior vena cava; IVC: inferior vena cava; PV: pulmonary veins; a: aconitine.
Results
The remifentanil dose with minimal hemodynamic effect in the barbiturate-anesthetized dog was of 0.5 mg/kg (group I; n = 5). During the temporal course recording of the digitalis intoxication (group II) it was found that digoxin (9 mg/kg/min) reached the LD (0.527 ± 0.079 mg/kg; n = 5) in an average time of 63.25 ± 11.3 min. This is consistent with the report published in 1964 in which, during the temporal course of digitalis intoxication, an A-V block is produced early and then continues until reaching ventricular fibrillation and asystolia.19 In group II, remifentanil given as a single bolus abolished the effects produced by digoxin in all cases, reverting the A-V dissociation and eliminating the multifocal ventricular extrasystoles, returning to a sinus rhythm and delaying the LD time to 100 ± 11.8 min.
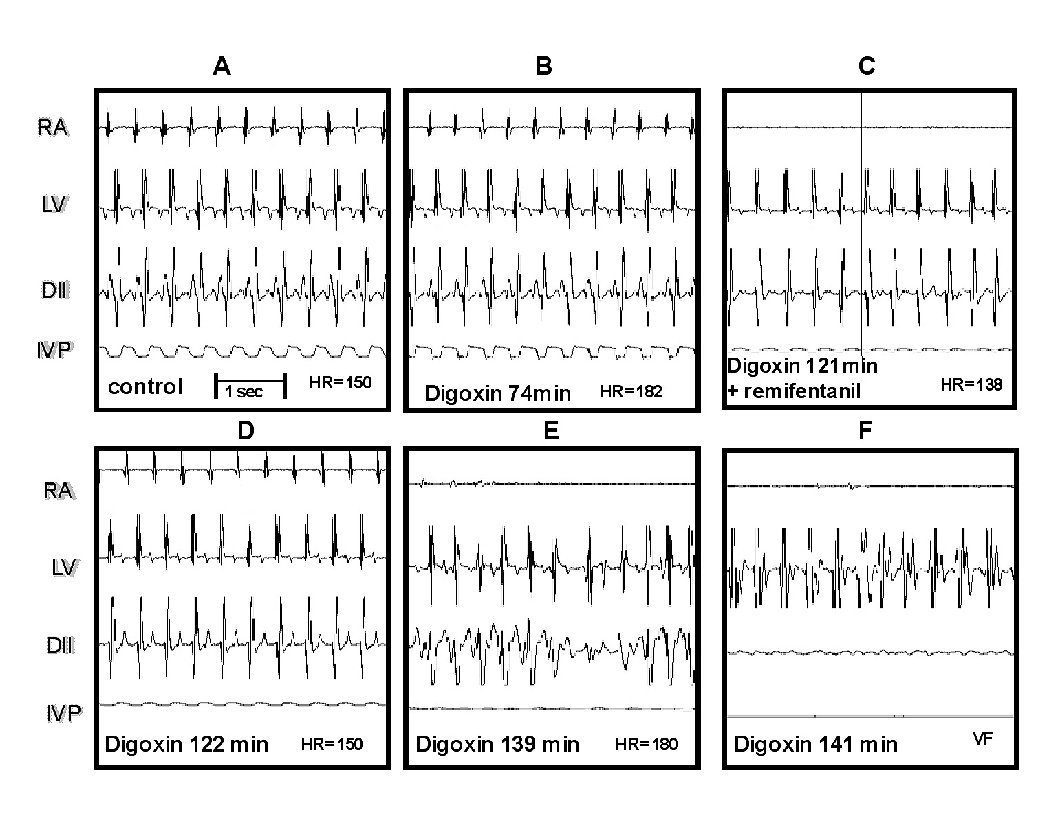
The Figure 2 illustrates these results. Panel A shows the control recordings. Panel B corresponds to the beginning of digoxin administration (74 min). Panel C, at 121 min of digitalis infusion, an atrial asystolia in right atrium (RA) can be observed, as well as absence of P wave in DII, and a drop of IVP, just before that remifentanil bolus was applied. Panel D ,1 min afterwards, depicts the remifentanil effect, observing again the trace in RA, the sinus rhythm is recovered in DII, and the HR returns to control values. E and F (18 and 20 min afterwards, respectively) depict the reestablishment of the digitalis intoxication course that continues until producing ventricular fibrillation and atrial and ventricular asystolia. When using two remifentanil boluses (n = 2), the LD time increased to 145 min in one dog; in the other dog, neither ventricular fibrillation nor death occurred even after 3 hours of recording and maintaining the digoxin infusion (electrocardiographic data not shown).

Figure 2. Effect of remifentanil on digitalis intoxication. Panel A, control tracings of the electrical activity recording of the right atrium (RA), the left ventricle (LV), the lead DII and the left intraventricular pressure (IVP).Panel B, electrocardiographic signs of digitalis intoxication.Panel C, atrial asystolia, at that moment remifentanil was administered. Panel D, the RA trace is recovered as well as the sinus rhythm in DII. Panels E and F, the digitalis intoxication follows its course leading to ventricular fibrillation (VF) at 141 min.
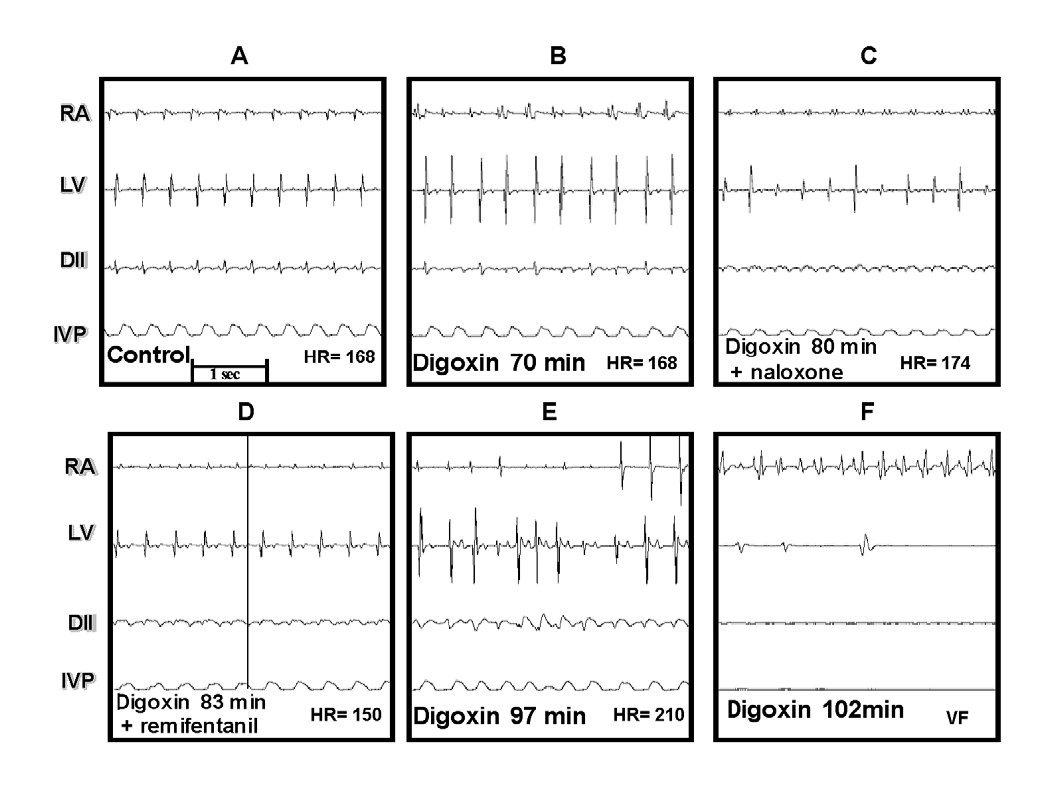
In group IV, once digoxin and naloxone were administered, remifentanil was not able to revert the digitalis intoxication. Figure 3 illustrates these effects. Panel A depicts the control electrocardiographic recording. Panel B depicts the first signs of the digoxin-induced intoxication (70 min.), C shows the atrial flutter, the irregular rhythm in left ventricular (LV), and ventricular fibrillation in DII; at this moment, naloxone was administered (80 min), 3 min later, remifentanil was administered in a single bolus, but it did not produce any appreciable effect on the digoxin-induced changes, as observed in E and F.

Figure 3. Effect of naloxone and remifentanil on digitalis intoxication. A, control. B, beginning of digitalis intoxication. C, naloxone is administered at 80 min, D, remifentanil is administered at 83 min. E and F, temporal course of digitalis intoxication similar to that shown in Figure 2.
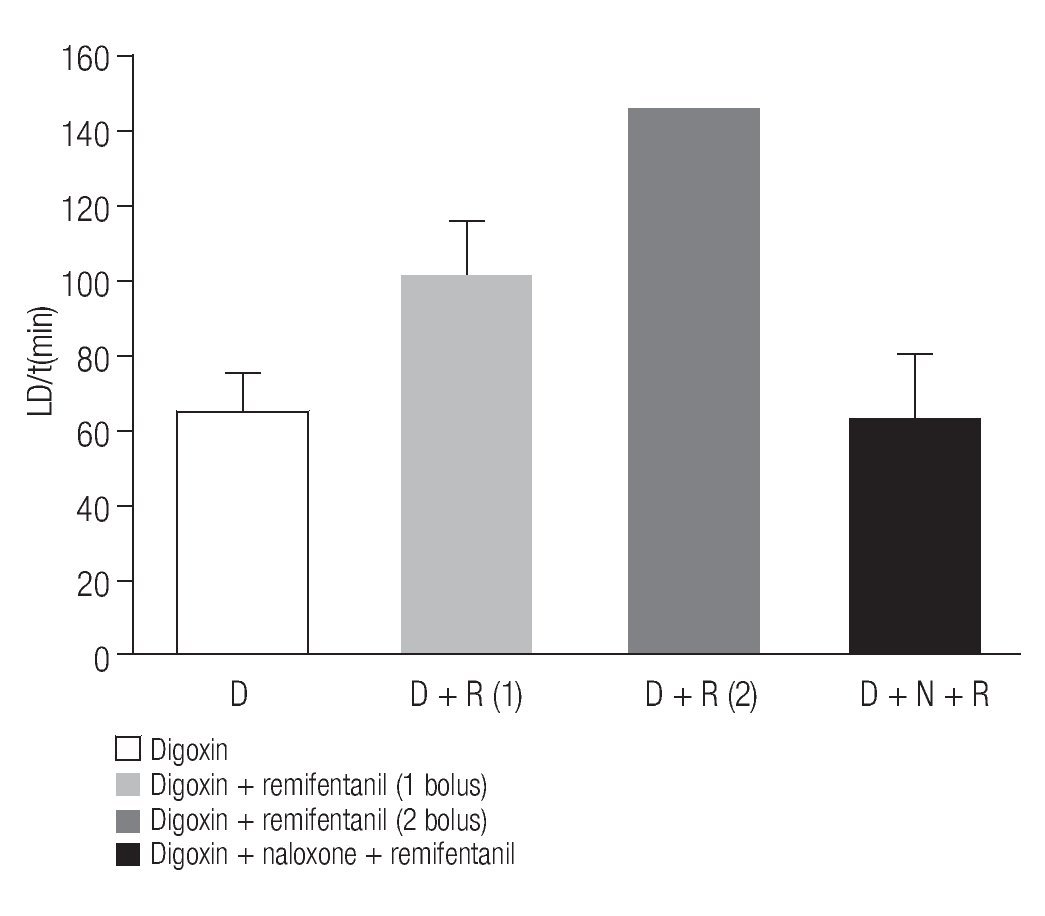
The average time LD (min) obtained in the previous groups is show in Figure 4. In the digitalis-induced intoxication group (D) the LD was of 63.25 ± 11.3 min (n = 5); in the digoxin plus remifentanil group (D + R), LD was of 100 ±11.8 min (n = 5). The third bar corresponds to the value obtained in a single animal (145 min) that received two boluses of remifentanil (D+R(2)). It is important to take in account that the other dog that also received the two remifentanil boluses, it was not considered in this graph. The last bar corresponds to the digoxin, naloxone, and remifentanil (D+N+R) group in which the LD was similar to that of the control group, 65.2±13.8 (n = 5).

Figure 4. Time needed to reach the LD of digoxin in the diverse groups. Average values of LD obtained in the digitalis intoxication group (D), of digoxin plus a remifentanil bolus (D + R), digitalis intoxication plus two remifentanil boluses (D + R(2)), and finally that of digoxin plus naloxone and remifentanil (D + N +R) are shown. Bars represent the average of 5 experiments, except in DR(2).
The Figure 5 illustrates one of the results obtained with group V, using the double arrhythmia model to study the effect of a single remifentanil bolus (0.5 mg/kg). Panel A shows the control recording. Panel B depicts the asystolia in the RAA area induced by its clamping, the other recordings remain practically stable. In C, the ectopic focus produced by aconitine in RAA can be seen as well as the start of the atrial flutter in RA. Once the double arrhythmia has been established, the remifentanil bolus is applied (panel D). Panel E shows the gradual disappearance of the ectopic focus and atrial flutter, as well as reversal to sinus rhythm in DII. In F it was not possible to maintain the antiarrhythmic effect in the aconitine-induced arrhythmia; however, it can be observed that the rest of the tracings remain close to the control tracing, although at a higher frequency. Remifentanil, in this experimental model, eliminated three of the five atrial tachycardias induced by aconitine and two of the five atrial flutters.

Figure 5. Effect of remifentanil on the double arrhythmia. A, control electrocardiographic recording showing the trace of the right atrial appendage (RAA), in the right atrium (RA), in lead DII (DII), and the recording of the left intraventricular pressure (IVP). B, asystolia in the RAA area induced by clamping, the remainder remains practically stable. C, ectopic focus in RAA induced by aconitine. Observe also the start of the atrial flutter in RA. D, corresponds to remifentanil bolus administration. E and F, note the gradual disappearance of the ectopic focus and atrial flutter; observe the reversion to sinus rhythm in DII.
Discussion
Opioids have been demonstrated to possess antiarrhythmic activity especially in those arrhythmias associated to an ischemia-reperfusion injury.1-6 Our results demonstrate the antiarrhythmic effect of remifentanil in arrhythmias induced by digitalis intoxication and in the double arrhythmia model in anesthetized dogs. The remifentanil dose used (with the minimal hemodynamic effect) was 0.5 μg/kg (group I). This value correlates with that reported by other authors, who measured the anesthetic potency of remifentanil in dogs anesthetized with enflurane, finding that a 0.72 mg/kg/min dose produces a 50% decrease in the minimal alveolar concentration (MAC).20 This remifentanil dose abolished the A-V dissociation and the ventricular extrasystoles, reverting to a sinus rhythm in all animals of the digitalis intoxication group (group III). This effect seems to be mediated by opioid receptors since naloxone was able to inhibit the antiarrhythmic effect of remifentanil, as observed in the presence of this antagonist. To determine the type receptors involved in these effects more specific studies are needed.
In the double arrhythmia model, remifentanil presented an interesting effect on the ectopic focus although transient. Besides, it only eliminated three of the produced ectopic foci. Remifentanil exerted a lower effect on the circular movement although more prolonged in time, since it was maintained until the last stage of recording. This could be due to an increase in the refractory period induced by remifentanil, being enough to prolong the wavelength. Likewise, remifentanil presented a partial antiarrhythmic effect, as it eliminated only two of the five induced atrial flutters. In summary, in our experimental conditions, we observed a 59% and 129% (the latter in only one case; (Figure 4) increase in the LD of digoxin induced by remifentanil, plus one case in which it was not possible to maintain the digitalis intoxication toward fibrillation and death using a double remifentanil dose. These data suggest that remifentanil is capable of activating dose-dependent myocardial protective effects; this suggestion requires additional research for its confirmation. The antiarrhythmic effects of remifentanil could be related to changes in the ionic channels activity of the myocardium. In this sense, a morphine-induced increase in duration of the cardiac action potential and hyperpolarization of the membrane resting potential, due to an increase in the potassium current IK1, in rabbit ventricular myocytes have been reported.21 Currently, there is no consensus regarding the mechanism of opioids involved in the induction of cardioprotection and increase in the resistance to arrhythmogenic factors; however it seems that rather diverse mechanisms are involved. Some authors suggest that the autonomous nervous system –in its sympathetic branch- plays an important role in this mechanism.22 Other authors attribute the beneficial effects of opioids to activation of opioid receptors of the myocardium that inhibit adenylcyclase activity with the consequent decrease in cAMP production.11 This would lead to a negative effect in cardiac inotropism and chronotropism.
On the other hand, it has been proposed that activation of opioid receptors stimulates the production of nitric oxide, which could contribute to the electrical stability of the heart acting on the endothelium of the coronary vessels and increasing blood flow to the myocardium.23 It has also been reported that opioid peptides increase the resistance not only to ischemia-reperfusion-induced arrhythmias but also to catecholamine- and aconitine-induced arrhythmias.24 There are other arrhythmogenic mechanisms on which the antiarrhythmic activity of opioids has not been tested, such as arrhythmias due to atrial flutter re-entry and the different types of arrhythmia produced by digitalis intoxication. In the present study, we found that remifentanil has an antiarrhythmic effect on arrhythmias induced in the atrium (double arrhythmia model) as well as on the ventricular arrhyhtmia produced by digitalis intoxication.
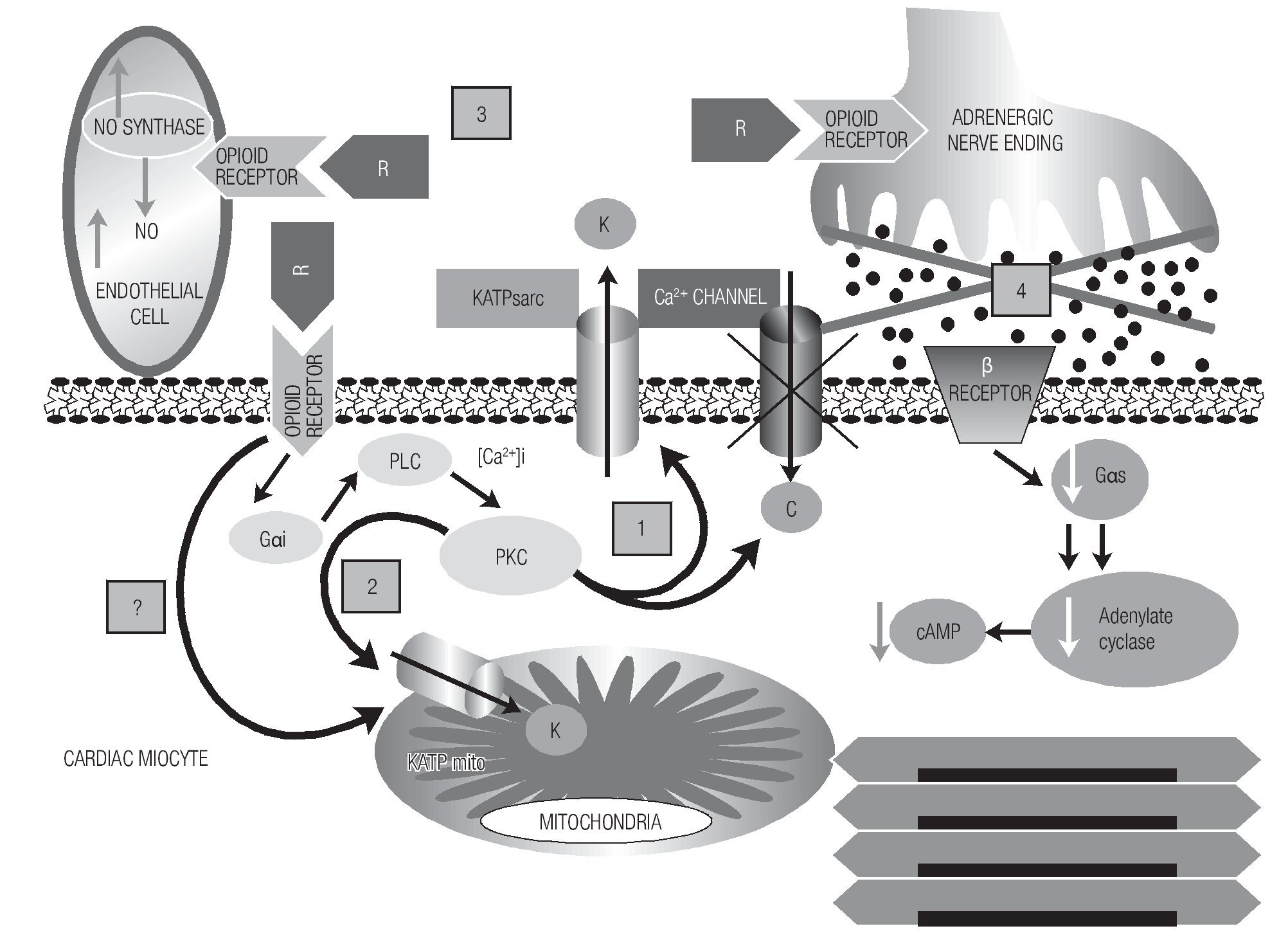
Most opioid agonists show affinity to a certain type of opioid receptor. Remifentanil is mainly a mu-agonist but it also exerts an effect on delta and kappa receptors.14 Opioid receptors are present in practically all organs and tissues but differ in the distribution of the subtypes. In the human heart, there are three subtypes distributed in the atrium and in the ventricle at different proportions.25 Therefore, activation of opioid receptors can induce heart resistance to the cardiotoxic and arrhythmogenic effect of diverse pharmacological agents. According to our results, probably the highest resistance to digitalis intoxication, the partial reversal to digoxin intoxication, and the antiarrhythmic activity induced by remifentanil resulted from the action of the opioidergic system, mainly on the opening of the sarcolemal and mitochondrial KATP channels, aside from the simultaneous inhibition of the glycogen-synthase-kinase 3-beta, as proposed by other authors.26 Lishmanov et al. suggest that activation of opioidergic receptors exerts antiarrhythmic action against aconitine-induced arrhythmias by inhibiting the sodium-calcium (Na/Ca) exchange,5 we could add to this mechanism the action of opioids exerted on the KATP channels. This effect could explain one of the electrophysiological conditions to suppress re-entry phenomena, as is the prolongation of wavelength postulated by Rosenblueth.17 The cardioprotective and antiarrhythmic effect of opioids could be mediated by diverse intracellular signaling pathways and by indirect effects as summarized in Figure 6.

Figure 6. A diagram of the possible mechanisms by which remifentanil (R) could exert its antiarrhythmic and cardioprotective effects. Mechanisms are shown with numbers. After the binding of the opioid agonist to its receptor in the myocyte, phospholipase C is activated by means of regulatory G-proteins, which through second messengers (DAG, IP3, increase in [Ca2+]i) activates PKC, in turn, this triggers several cellular responses. One of them is the blockade of Ca2+ channels and opening of the sarcolemal KATP channel [1]. Mechanism [2], KATP mito channels are also activated, one part dependent on the PKC-signaling pathway; however, opioidergic receptors could exist in the mitochondrial membrane that, when stimulated, could directly activate these channels. This possibility is marked with [?] sign. Mechanism [3]): as an indirect mechanism, activation of coronary endothelial receptors that could stimulate the nitric oxide synthase; this, in turn, increases the hyperhemic index of the heart, contributing to the electrophysiological stabilization of the myocardium against arrhythmias. Mechanism [4] is another indirect one, in which the binding of opioids to their receptors in the adrenergic terminals would produce a sympathetic inhibition that would reduce the intramyocardial cAMP.
Aside from these mechanisms, other less characterized ones have been proposed to participate in the opioid effects, such as release of adenosine induced by opioids and the production of nitric oxide in the cardiac myocyte.23
Clinical implications
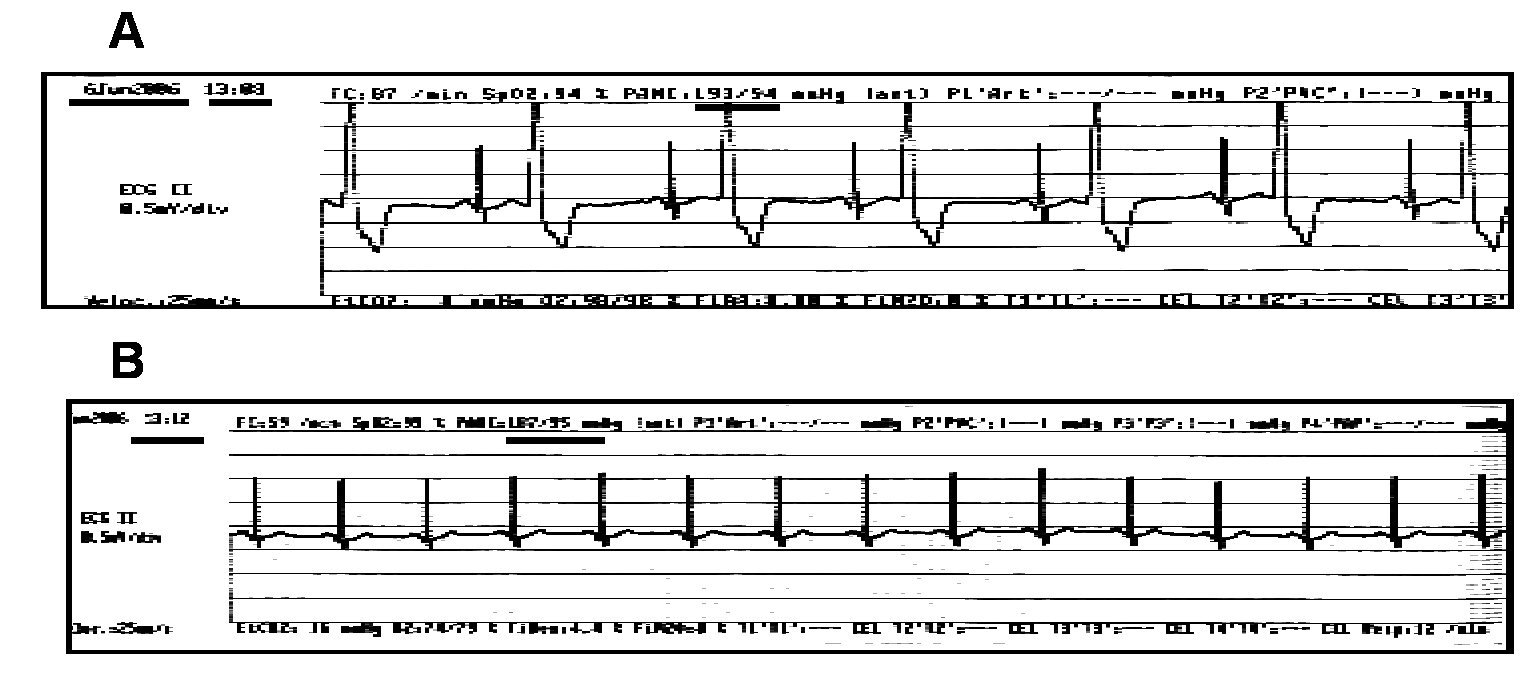
Opioids are widely used as analgesic in intensive care units and during surgery as part of the anesthesia for cardiac and non-cardiac surgeries. Remifentanil is preferred for its rapid onset and the fast recovery from anesthesia. Recently, it has been reported that remifentanil avoided prolongation of the QTc interval associated to tracheal intubation during anesthesia induction with sevoflurane.27 Figure 7 depicts the case of a patient with digitalis intoxication, in whom remifentanil infusion at 0.15 μg/kg/min reverted the characteristic bigeminism induced by this intoxication.

Figure 7. Effect of remifentanil in a patient with digitalis intoxication. Male patient, 64-year-old, weight of 100 kg. He was undergoing to right knee arthroscopy. He had been receiving Lanoxin 0.125 mg daily for several years, Angiotrofin 180 mg one single daily dose, Aspirin 300 mg daily, the latter was suspended one week before surgery. The pre-surgery ECG revealed blockade of the right branch of the Hiss bundle (RBBB) and paroxistic tachycardia, as well as ischemic heart disease; the patient had been implanted three coronary stents in 1994. The upper part, A, of the figure depicts the pre-surgery ECG in which bigeminism was observed. Part B reveals the absence of bigeminism at 4 min after starting remifentanil infusion at 0.15 μg/kg/min.
These clinical findings, together with our results, justify the use of opioids for cardioprotection during pre- and post-conditioning of ischemia and for the prevention of myocardial damage induced by ischemia-reperfusion in patients subjected to coronary angioplasties and to coronary arteries revascularization surgery.28,29
Corresponding author:
Gustavo Pastelín,
Departamento de Farmacología, Instituto Nacional de Cardiología “Ignacio Chávez”, Juan Badiano No. 1, Col.
Sección XVI, México, D.F. CP 14080
Received: December 10, 2008;
accepted: July 27, 2009.