


La enfermedad cardiovascular representa la primera causa de morbimortalidad a nivel mundial. Actualmente, la evidencia que sustenta la implementación de determinadas intervenciones terapéuticas se origina a partir de datos provenientes de grupos poblacionales. Sin embargo, los pacientes presentan variaciones interindividuales relacionadas tanto con la eficacia como con la toxicidad ante un mismo tratamiento farmacológico. Estas variaciones pueden ser explicadas principalmente por diferencias en la adherencia, interacciones no reconocidas y diferencias genéticas. Las alteraciones en el genoma explican entre un 20 y un 95% de la variabilidad interindividual tanto en la disponibilidad como en la respuesta a fármacos. En el tratamiento de las enfermedades cardiovasculares existen diversos ejemplos de dicha variabilidad genética interindividual y su impacto en la eficacia o toxicidad de diferentes fármacos. La variabilidad genética que determina la respuesta al clopidogrel radica fundamentalmente en el polimorfismo del citocromo (CYP) 2C19. Los polimorfismos en los genes CYP 2C9 y VKORC1 explican gran parte de la variabilidad en la respuesta a los anticoagulantes dicumarínicos. Con respecto al tratamiento hipolipidemiante, el polimorfismo del gen SLCO1B1 se ha asociado a la aparición de miopatía en pacientes tratados con simvastatina. Muchos otros polimorfismos han sido postulados pero sin un impacto clínico definido hasta la fecha. La utilización de la farmacogenómica en la práctica cotidiana ofrece la oportunidad de poder predecir toxicidad o eficacia terapéutica.
Cardiovascular disease remains a major cause of morbidity and mortality worldwide. Current medical practice takes into account information based on population studies and benefits observed in large populations or cohorts. However, individual patients present great differences in both toxicity and clinical efficacy that can be explained by variations in adherence, unknown drug to drug interactions and genetic variability. The latter seems to explain from 20% up to 95% of patient to patient variability. Treating patients with cardiovascular disorders faces the clinician with the challenge to include genomic analysis into daily practice. There are several examples within cardiovascular disease of treatments that can vary in toxicity or clinical usefulness based on genetic changes. One of the main factors affecting the efficacy of Clopidogrel is the phenotype associated with polymorphisms in the gene CYP 2C9. Furthermore, regarding oral anticoagulants, changes in CYP2C9 and VKORC1 play an important role in changing the clinical response to anticoagulation. When analyzing statin treatment, one of their main toxicities (myopathy) can be predicted by the SLCO1B1 polymorphism. The potential for prediction of toxicity and clinical efficacy from the use of genetic analysis warrants further studies aiming towards its inclusion in daily clinical practice.
Muchos de los progresos terapéuticos de la medicina moderna se sustentan en un mayor conocimiento acerca de la biología de las enfermedades y sus mecanismos fisiopatogénicos. Paralelamente, resulta evidente que la mayoría de los fármacos en sus dosificaciones convencionales no producen efectos uniformes en todos los pacientes que los reciben1. Los fármacos prescritos para las indicaciones más frecuentes en la práctica habitual no producen el efecto terapéutico deseado en el 30 al 60% de los enfermos2.
Mientras los grandes ensayos aleatorizados evalúan el beneficio de distintas estrategias a nivel grupal, los pacientes presentan variaciones interindividuales tanto en la eficacia como en la toxicidad. Dichas variaciones se pueden explicar principalmente por diferencias en la adherencia, interacciones no reconocidas y por diferencias genéticas. Varios fármacos útiles en el tratamiento cardiovascular presentan límites terapéuticos estrechos y diferencias en la efectividad debido a variaciones genéticas. Se considera que las variaciones en el genoma explicarían entre un 20 y un 95% de la diferencia interindividual tanto en la disponibilidad como en la respuesta a fármacos3.
La farmacogenómica es el estudio de la influencia de las variaciones genéticas heredadas o adquiridas en la respuesta a fármacos4. Los efectos farmacológicos de la mayoría de los medicamentos habitualmente prescritos son la resultante de una serie de procesos farmacocinéticos (que determinarán la cantidad de medicamento que llega a la biofase) y farmacodinámicos vinculados a la efectividad de la interacción del fármaco con su receptor. La variabilidad de expresión y función de las distintas enzimas involucradas en estos procesos constituye el núcleo principal de estudio de la farmacogenómica. Es claro que este análisis no explica la totalidad de la variabilidad de la respuesta terapéutica, sino que se enmarca dentro de las diferencias determinadas por género, edad, alimentación, comorbilidades, factores ambientales e interacciones de medicamentos. Asimismo, la farmacogenómica puede facilitar la identificación de biomarcadores útiles para la selección del fármaco adecuado, la dosis apropiada, el tiempo de tratamiento óptimo o para la prevención de la aparición de efectos adversos2.
Existen distintos tipos de variantes genéticas posibles (deleciones, inserciones o multiplicaciones), que pueden involucrar porciones relativamente extensas del ácido desoxirribonucleico (ADN) celular, aunque las más frecuentes y blanco habitual de los análisis farmacogenéticos son los polimorfismos de nucleótido único (single nucleotide polymorphism o SNP según sus siglas en inglés). La secuenciación del genoma humano ha permitido establecer que hay más de 10 millones de SNP, sitios específicos en los que existe un cambio de la secuencia de nucleótidos. Entre ellos solo una minoría parece tener algún tipo de impacto en la cinética o la dinámica de los fármacos. Por eso, las etapas del estudio farmacogenómico pueden ir desde el aspecto puramente genético hacia la identificación de los SNP con impacto clínico o bien, en sentido inverso, estableciendo las secuencias específicas de nucleótidos, a partir de la identificación de individuos con comportamientos particulares en relación con el metabolismo de fármacos5,6. Los genes que codifican las proteínas metabolizadoras, transportadores o receptores, pueden presentar distintas variantes alélicas, algunas de las cuales demuestran un impacto diverso sobre la magnitud de la expresión de sus productos7.
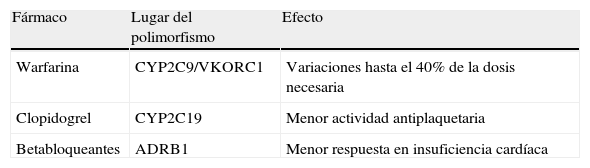
En la última década, el conocimiento de la farmacogenómica relativa a los fármacos cardiovasculares se ha incrementado significativamente. A continuación, describimos las principales aplicaciones de la farmacogenómica en enfermedades cardiovasculares, en lo referente a fármacos con acciones muy diversas (tabla 1). Existen procedimientos avanzados como la guía farmacogenómica del tratamiento anticoagulante con agentes dicumarínicos, mientras otros buscan definir su lugar en la terapéutica como la farmacogenómica del clopidogrel y las estatinas. Finalmente, en un estadio más incipiente, se encuentran los betabloqueantes, otros antihipertensivos y algunos agentes antiplaquetarios.
ClopidogrelEl clopidogrel inhibe la activación plaquetaria inducida por la adenosina difosfato mediante la unión irreversible al P2Y12, receptor plaquetario de la adenosina difosfato. De este modo impide la agregación plaquetaria8. Existe una gran variabilidad interindividual en la respuesta al clopidogrel entre pacientes que reciben el fármaco luego de una intervención coronaria percutánea. El clopidogrel es un profármaco, que requiere activación metabólica en pasos secuenciales, con la isoenzima CYP2C19 como paso limitante. Su gen codificante es altamente polimórfico, con un SNP asociado a la aparición de una proteína con muy escasa actividad enzimática9. La biodisponibilidad del clopidogrel está limitada por la actividad de la glucoproteína P intestinal, codificada por el gen ABCB1, y solamente un 15% de la dosis original es oxidada a su metabolito activo10.
La presencia de alelos variantes en el gen que codifica a la isoenzima CYP2C19 está asociada a una escasa actividad enzimática; en particular el alelo *2, y en menor medida el *3, *4 y *5, se asocian a una disminución de la respuesta plaquetaria al clopidogrel11–13. Si bien persisten ciertas controversias respecto a la metodología más apropiada para evaluar la respuesta plaquetaria al uso de clopidogrel, este fenómeno se ha asociado con una mayor actividad plaquetaria residual y una menor respuesta terapéutica en pacientes con enfermedad cardiovascular, luego de recibir este fármaco en dosis de carga10,14–17. Por otro lado, la presencia del alelo *17 se asocia a un incremento de la actividad transcripcional, y por consiguiente, a una mayor susceptibilidad a la acción del clopidogrel con incremento del riesgo de sangrado18.
La variabilidad genética que determina la respuesta al clopidogrel no se limita solamente a los polimorfismos de CYP2C19. La influencia de otros polimorfismos, tales como los del gen ABCB1 o los del receptor P2Y12, no ha sido informada de manera consistente en distintas publicaciones15,19–21. En 2010 la Food and Drug Administration incluyó una advertencia en el prospecto del producto. Este señala que las personas con una variante disfuncional en el gen codificante de CYP2C19 pueden requerir ajustes de la dosis de clopidogrel o el uso de un fármaco alternativo22. Se ha establecido la utilidad de la genotipificación de CYP2C19 para pacientes con riesgo moderado o alto de eventos cardiovasculares, que sean tratados con clopidogrel23,24.
La evidencia de la asociación con eventos clínicos permanece aún controvertida. En 2 ensayos clínicos de gran magnitud, no pudo ser demostrada la asociación de los polimorfismos del gen CYP2C19 con el riesgo de eventos cardiovasculares en pacientes con episodios coronarios agudos o fibrilación auricular. Lo mismo aconteció en una revisión sistemática que incluía a pacientes con distintas indicaciones para recibir clopidogrel24–26. Un estudio clínico realizado recientemente en Japón sobre 160 pacientes, con intervención coronaria percutánea, demostró que el riesgo de eventos adversos cardíacos importantes y de necesidad de revascularización posterior fue significativamente mayor entre pacientes con fenotipo de metabolizadores escasos e intermedios para CYP1C19 respecto a los metabolizadores grandes27. Por otro lado, el análisis farmacogenómico del estudio Pharmacogenomics of Antiplatelet Intervention (PAPI) realizado sobre 429 personas de descendencia Amish, demostró que la presencia de la variante *2 en el gen CYP2C19 estaba asociada con una disminución de la respuesta de agregación plaquetaria al clopidogrel y con un riesgo aumentado (hazard ratio 2.42; IC95%: 1.18-4.99), de padecer un evento cardiovascular isquémico o fallecer dentro del año del seguimiento28. Finalmente, en un metaanálisis más reciente basado en 9 estudios farmacogenómicos en 9,685 pacientes con eventos coronarios agudos o que fueron sometidos a intervención coronaria percutánea, la presencia de alelos variantes asociados con expresión proteica defectuosa, tanto en homo como en heterocigosis, estaba ligada a un mayor riesgo de muerte por causa cardiovascular, infarto agudo de miocardio o accidente cerebrovascular29.
Estos hallazgos muestran que el análisis farmacogenómico para el clopidogrel podría ser de utilidad en pacientes seleccionados, como los que son sometidos a intervenciones coronarias percutáneas y presentan mayor riesgo de trombosis30. En estos escenarios, un aspecto crucial para la demostración de la utilidad del estudio farmacogenómico es el tiempo de respuesta. La disponibilidad de un análisis en la «cabecera» del paciente con resultado rápido o inmediato mejoraría significativamente la perspectiva con relación a la individualización terapéutica. Esto ha sido confirmado recientemente31.
Otros antiagregantes plaquetariosLos antiplaquetarios representan parte del estándar de cuidado en el tratamiento del síndrome coronario agudo y en la prevención primaria y secundaria de eventos cardiovasculares. Existen marcadas variaciones individuales en la acción antiplaquetaria de tales fármacos y los análisis cuantitativos han demostrado una distribución prácticamente normal. A día de hoy, hay abundante evidencia demostrativa de una base genética tanto para la variabilidad en la acción plaquetaria como en la respuesta al tratamiento antiplaquetario. En primer lugar, existen varios polimorfismos asociados a variaciones en la función plaquetaria basal. Diferencias en las glucoproteínas ia-iia, ib-ix y vi han sido relacionadas con cambios en la adhesión plaquetaria. Más aún, variantes en el receptor P2Y, PAR 1 y el receptor de tromboxano A2 modifican la capacidad de las plaquetas de activarse y agregarse32,33.
La aspirina inhibe irreversiblemente la ciclooxigenasa 1 plaquetaria, enzima que cataliza la transformación de ácido araquidónico en prostaglandina 2, convertida a tromboxano A2. Numerosos receptores plaquetarios y sus polimorfismos genéticos se han postulado como modificadores potenciales del efecto antiplaquetario de la aspirina; sin embargo, su impacto clínico no está bien determinado. Polimorfismos en la ciclooxigenasa 1, en el receptor P2Y1 y en la glucoproteína ia-iia han sido descritos pero sin impacto clínico evidente34. En un reciente metaanálisis, el único asociado a resistencia al tratamiento con aspirina fue un polimorfismo frecuente de la glucoproteína iiia (OR: 2.49), sin un claro impacto en eventos clínicos relevantes.
Con respecto a los inhibidores de la glucoproteína iib-iiia, existe evidencia de una alta tasa de variabilidad interindividual en la inhibición de la agregación plaquetaria. Una variante polimórfica de iiia (HPA1-b) se asocia a menor efecto antiagregante del abciximab, sin un claro efecto clínico a pesar de estar asociado en algunas cohortes con menor riesgo de sangrado y mayor incidencia de recurrencia de eventos trombóticos35,36.
Anticoagulantes orales dicumarínicosLos anticoagulantes orales dicumarínicos (acenocumarol y warfarina) son fármacos muy utilizados para la prevención y el tratamiento de accidentes cerebrovasculares cardioembólicos, trombosis venosa y como parte del manejo terapéutico de la fibrilación auricular. Los dicumarínicos son fármacos de muy estrecho intervalo terapéutico. El 33% de las hospitalizaciones en EE. UU., secundarias a efectos adversos relacionados con fármacos, fueron debidas a estos anticoagulantes orales37. Tal riesgo es mayor durante las primeras 4 a 8 semanas de iniciada la anticoagulación38. Por eso resulta crítico lograr una dosis segura y efectiva desde el inicio de la anticoagulación. Una particularidad de estos agentes es la gran variabilidad interindividual en la respuesta, la cual puede explicarse en parte por la edad, las interacciones de medicamentos, infecciones, disponibilidad de vitamina K en la dieta, función cardíaca, alteración en la función hepática y también por la presencia de polimorfismos genéticos39,40.
Los genes CYP 2C9 y VKORC1 parecen ser los únicos con efectos relevantes en la respuesta a los anticoagulantes dicumarínicos40. La acenocumarina comercialmente disponible es una mezcla racémica, en la cual el R-enantiómero es el más importante desde el punto de vista farmacológico, debido a que su vida media es de 8h, mientras que la del S-enantiómero es de 2h. Ambos enantiómeros son metabolizados a 6 y 7 hidroxiderivados primariamente a través de la isoenzima CYP2C9 del complejo CYP450, y secundariamente a través de CYP1A2 y CYP2C19. Si bien la gran mayoría de los estudios farmacogenómicos publicados han evaluado la warfarina, dada la similitud de sus características farmacológicas, se estima que la variabilidad genética del CYP2C9 determina el 14% de la variabilidad interindividual de la respuesta de los pacientes a la acenocumarina41. La isoenzima CYP2C9 es codificada por el gen CYP2C9, el cual presenta variabilidad genética. In vivo, 2 SNP de CYP2C9 (alelos *2 y *3) se han asociado con menor expresión de la isoenzima y con aumento de la respuesta a los anticoagulantes orales (23 veces mayor riesgo de sangrado durante la inducción).
Tanto la warfarina como la acenocumarina actúan inhibiendo la enzima vitamina K epóxido reductasa (denominada VKORC1 por vitamina K epóxido reductasa cómplex, la cual es codificada por el gen VKORC1) que regenera la vitamina K reducida, la cual a su vez actúa como cofactor de la gammacarboxilasa, enzima que activa los factores de la coagulación y otras proteínas K dependientes (ii, vii, ix y x, proteína C, S y Z) mediante la carboxilación postranscripcional de sus residuos de ácido glutámico.
VKORC1 es el blanco de acción de los anticoagulantes dicumarínicos, y su gen codificante VKORC1 presenta distintas variantes polimórficas en desequilibrio de ligamento. Las variantes con importancia funcional dentro del promotor y de la región intrónica del gen son 1639G→A o 3673, 497 T→G o 5808, 1173 C→T o 6484, 1542 G→C o 6853, 2255 C→T o 7566. Dada su herencia combinada, la identificación de cualquiera de estos polimorfismos permite la caracterización del paciente como haplotipo A que presenta mayor sensibilidad a la acción de dichos fármacos42.
Dado que el mayor riesgo de efectos adversos (especialmente hemorragias) se establece dentro de las primeras 8 semanas de iniciado el tratamiento anticoagulante, y que, con el manejo actual, solamente del 45 al 64% de los pacientes logra un international normalized ratio (INR) terapéutico dentro de ese período, el conocimiento farmacogenómico podría permitir la selección de una dosis personalizada de acenocumarina para el inicio del tratamiento anticoagulante41–43. Si bien los anticoagulantes orales que se utilizan en la actualidad podrían ser reemplazados en el futuro cercano por otros medicamentos más modernos como los inhibidores directos de la trombina y los inhibidores del factor x, los dicumarínicos siguen siendo los fármacos de elección para la anticoagulación, al menos por su conveniente costo-efectividad, así como la existencia de antídoto ante una eventual toxicidad y su eficacia históricamente comprobada40,41. El análisis farmacogenómico para warfarina ha demostrado ser de utilidad no solamente en el logro más rápido y persistente de niveles de INR42, sino también en la prevención de eventos clínicos (hospitalización por cualquier causa, por trombosis o hemorragia)44–46. A partir de estos resultados en EE. UU., la Food and Drug Administration ha reconocido el valor de la realización de las pruebas farmacogenómicas previamente a la utilización de fármacos anticoagulantes orales. La evidencia actual internacional demuestra que la individualización de la dosis de warfarina, guiada por estudio farmacogenómico, determina no solamente un beneficio en los valores de INR, sino también una reducción del riesgo de eventos clínicos adversos (rehospitalización y mortalidad por hemorragia o trombosis)44. Si bien se conoce que la acenocumarina comparte tanto la vía metabólica como el blanco de acción farmacológica con la warfarina, no se han realizado hasta el momento estudios clínicos que permitan analizar la asociación entre los polimorfismos de CYP2C9 y VKORC1 con los niveles de INR en pacientes tratados con este fármaco.
AntihipertensivosLos inhibidores de la enzima convertidora de angiotensina (ECA) constituyen un grupo farmacológico de primera línea en el tratamiento actual de la hipertensión arterial. Un polimorfismo de inserción-deleción (rs4646994) se asocia significativamente con la concentración plasmática de la ECA, aunque la utilidad de esta determinación no ha sido confirmada en ensayos clínicos47,48. Por otra parte, la mutación M235T del gen AGT, que codifica el angiotensinógeno, se ha asociado con el riesgo de accidentes cerebrovasculares y de infarto agudo de miocardio en pacientes que reciben inhibidores de la ECA, y su relevancia deberá ser confirmada en estudios posteriores49,50. Una combinación de 3 polimorfismos (2 en el gen del receptor tipo i de angiotensina ii y uno en el gen BKI del receptor tipo i de bradicinina) permitió predecir adecuadamente la respuesta y el riesgo de toxicidad en el uso de perindopril51.
Para el caso de los betabloqueantes, el polimorfismo Ser49Gly del receptor ß-1 se asoció con incremento del proceso de «downregulation» del receptor y permitió identificar a pacientes con miocardiopatía dilatada con mayor riesgo de mortalidad a 5 años de tratamiento con bajas dosis52,53. El polimorfismo Arg389Gly del mismo receptor presente en homocigosis se asoció significativamente con una mejoría de la fracción de eyección, una reducción del riesgo de hospitalización y de mortalidad, en pacientes tratados con betabloqueantes52,54. El impacto de la presencia de este polimorfismo sobre la reducción de la tensión arterial es controvertido.
Los polimorfismos y mutaciones genéticas fuertemente asociados con los efectos de fármacos antihipertensivos aún no se han podido determinar. La mayoría de los resultados obtenidos de estudios farmacogenómicos o farmacogenéticos no han sido validados o no se han podido replicar en otros estudios55.
EstatinasLas estatinas son inhibidores de la enzima HMG CoA reductasa, paso limitante de la síntesis del colesterol. Son útiles para la disminución del colesterol LDL y la prevención primaria y secundaria de eventos cardiovasculares.
Diferentes polimorfismos en la HMG CoA reductasa y en el receptor de LDL han sido asociados con menores descensos en los niveles de LDL pero sin impacto clínico al menos evidente. El escenario más promisorio con respecto a las estatinas es la predicción de aparición de miopatía basada en la presencia de un polimorfismo del gen que codifica al OATP1B1, el polipéptido transportador de aniones orgánicos a través de la membrana hepatocitaria, responsable del ingreso de las estatinas en el hepatocito. Representa asimismo uno de los sitios de interacción entre las estatinas y los fibratos. El polimorfismo del gen SLCO1B1 (cromosoma 12) del transportador ha demostrado una asociación con la aparición de miopatía en subestudios de los ensayos SEARCH y HPS. En los pacientes aleatorizados con 80mg de simvastatina, la presencia de dicho polimorfismo se asoció a miopatía con un OR de 2756. Este escenario plantea la interesante opción de buscar dicho polimorfismo en los pacientes y ajustar posteriormente la dosis o el fármaco para disminuir la aparición de toxicidad. De todas formas, a pesar del aumento de riesgo, no hay que perder de vista la infrecuencia de miopatía (<1%) en los estudios clínicos en el momento de evaluar el impacto potencial en la práctica clínica.
En adición, el haplotipo H7 de la HmG CoA reductasa se ha asociado a menor respuesta al tratamiento con pravastatina y simvastatina, hallazgo que no se ha repetido para otras estatinas de uso común. Finalmente, la variabilidad en el control del colesterol ha sido asociada débilmente con variantes en la ApoE pero sin confirmaciones posteriores57,58.
Comentarios finalesLa mayoría de los desarrollos farmacogenómicos en terapéutica cardiovascular se encuentran actualmente en una fase de validación y demostración de su utilidad clínica potencial.
Las dificultades, comunes a los estudios genómicos para diversas áreas terapéuticas, incluyen problemas en el diseño original de los estudios, la inclusión de la farmacogenómica como objetivo secundario con el riesgo consiguiente de tamaños muestrales insuficientes, limitación en la repetición de resultados y la heterogeneidad fenotípica subyacente en la población59. Los avances significativos en este camino irán de la mano de la utilización de técnicas de secuenciación de nueva generación con tiempos de respuesta más rápidos, mayor claridad y focalización en las definiciones fenotípicas y las colaboraciones multicéntricas.
Se ha mostrado que la importancia relativa del análisis farmacogenético en la explicación de la variabilidad de respuesta a fármacos cardiovasculares no es suficiente como para justificar su incorporación en la práctica cotidiana60. Sin embargo, es importante recordar que para la gran mayoría de los fármacos ninguna variación individual es decisiva en cuanto a la predicción de respuesta, y esto es lógico desde un punto de vista evolutivo. Tal como ha sucedido en el caso de los anticoagulantes dicumarínicos, el análisis de las variaciones genéticas, en conjunto con aspectos clínicos individuales (tales como el peso, superficie corporal, edad, comorbilidades), incrementará la precisión de nuestras estimaciones.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses