



Introducción
La hipertensión arterial esencial (HTA, como se abordará en este texto) es una enfermedad multifactorial en la que participan factores ambientales, genéticos e intrínsecos como raza y género en su desarrollo.1-4 La HTA se define como una elevación de la presión sistólica igual o mayor a 140 mmHg, así como una elevación de la presión arterial diastólica igual o mayor de 90 mmHg. De hecho, un incremento incluso dentro del rango normal de la presión sistólica y diastólica tienen impacto continuo sobre enfermedades cardiovasculares.5,6 En 2005, la Organización Mundial de la Salud estimó que la mortalidad debida a enfermedades cardiovasculares fue de aproximadamente 17.5 millones a nivel mundial.5 En la Encuesta Nacional de Salud 2006 del Instituto Nacional de Salud Pública de México, se documentó la estimación de 17 millones de hipertensos en población adulta, cifra con la que se alcanza la prevalencia de 30.8%, que ubica a la HTA como una de las principales causas de morbi-mortalidad en nuestro país.7 Así, la HTA es el principal factor de riesgo para el desarrollo de infarto agudo al miocardio, falla cardiaca, arritmia ventricular, nefropatías, ceguera, entre otros serios problemas de salud. Aunque la HTA es una de las enfermedades más comunes a nivel mundial, poco se ha logrado en la comprensión sobre cuál o cuáles son los mecanismos moleculares y genéticos involucrados en su desarrollo. Sin embargo, en las recientes décadas se ha observado que el estilo de vida, sobrepeso, obesidad e inactividad física, en conjunto con otros factores como la ingesta elevada de sal en la dieta, adicción al alcohol, el género, entre otros que contribuyen drásticamente en el aumento de su prevalencia.5,8-10 No obstante, la HTA involucra otros eventos muy importantes en su fisiopatogenia, como la disfunción endotelial, el proceso inflamatorio y los factores genéticos.
Se ha estimado que el componente genético es muy importante para su desarrollo, de hecho varios estudios de heredabilidad muestran que entre 30% a 60% depende del componente genético para que se desarrolle HTA.1,2-11 Recientemente, con el uso de la tecnología de análisis de variantes genéticas del genoma completo, se han identificado diferentes loci que confieren susceptibilidad a desarrollar HTA.
Objetivo
Discutir los trabajos de genética y genómica en HTA que se han realizado tanto a nivel mundial como en nuestro país; estos estudios permitirán identificar aquellos genes que participan en la etiopatogenia de esta enfermedad. De esta manera, en el futuro se pretende que la HTA se pueda prevenir y que estas herramientas moleculares nos ayuden a establecer el diagnóstico y pronóstico más certeros, convirtiendo así a la genómica en una herramienta para hacer una medicina más personalizada e individualizada.
Estrategias genéticas y genómicas en la búsqueda de genes involucrados en la susceptibilidad para desarrollar HTA
Los estudios en familias han detectado que el componente genético es muy importante en el desarrollo de la HTA; varias publicaciones han puesto en evidencia que los genes juegan un papel principal en su desarrollo; el carácter hereditario llega a exceder 50%.1-2,11,12 Otros estudios que apoyan estos datos, son los realizados en gemelos concordantes (monocigotos) y discordantes (dicigotos), con quienes se ha mostrado también que el componente genético está fuertemente ligado a la HTA.13 Se ha calculado que el riesgo relativo (λs) en pares de hermanos afectados es de 3.5 veces mayor riesgo para hipertensión que el λs de la enfermedad arterial isquémica (λs = 2).12 Actualmente, los estudios genéticos se están enfocando a buscar los diferentes genes que pueden participar en la etiopatología de la HTA, lo cual, en el futuro, podría tener fuertes implicaciones diagnósticas y pronósticas en esta patología multifactorial.
Existen dos estrategias principales para identificar genes de susceptibilidad a HTA en humanos: a) los análisis de ligamiento y, b) los análisis de asociación. Ambas estrategias han sido ampliamente usadas con el objetivo de identificar a los genes que causan HTA.14 Estas estrategias no son mutuamente excluyentes y cada una presenta ventajas y desventajas.
Análisis de ligamiento
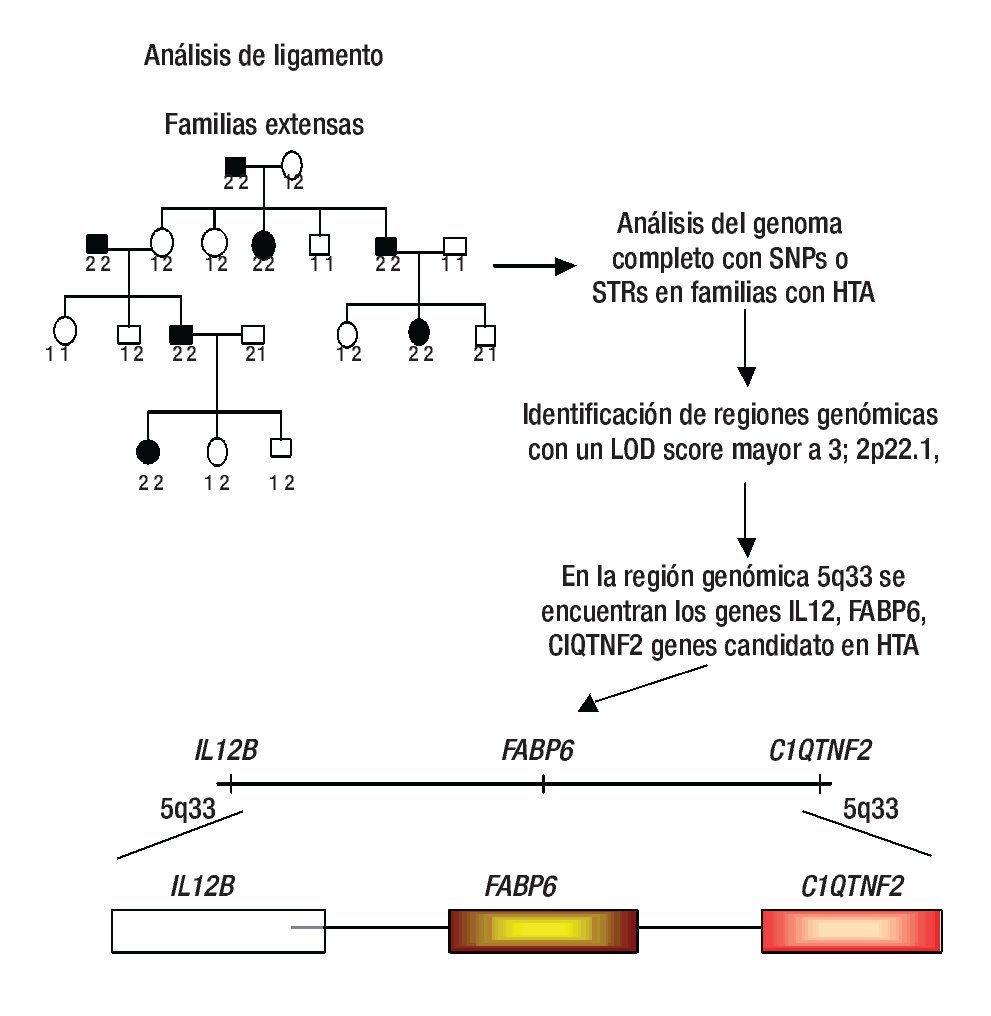
El ligamiento genético se refiere a la co-segregación de marcadores genéticos con el fenotipo de la enfermedad, por ejemplo HTA en familias con múltiples miembros afectados.14 De esta manera, se puede analizar la co-segregación de marcadores genéticos ampliamente distribuidos sobre todo el genoma, tales como los repetidos cortos en tándem (Short tandem repetitive: STRs) y los polimorfismos de un sólo nucleótido (single nucleotide polymorphisms: SNPs) con HTA; finalmente, se identifican los loci que contienen marcadores genéticos que son cosegregados junto con la HTA en diferentes familias (Figura 1).14,15 En los últimos años, en los análisis clásicos de escaneo del ligamiento del genoma completo (Genome-wide likage scan: GWLS) se utilizaban marcadores genéticos tipo STRs, los cuales tienen la característica de ser muy informativos; sin embargo, su distribución dentro del genoma no es tan amplia como los SNPs (se ha calculado que existen entre 10 y 20 millones), hasta hoy no hay estudios de ligamiento con este tipo de polimorfismos en HTA.16-18
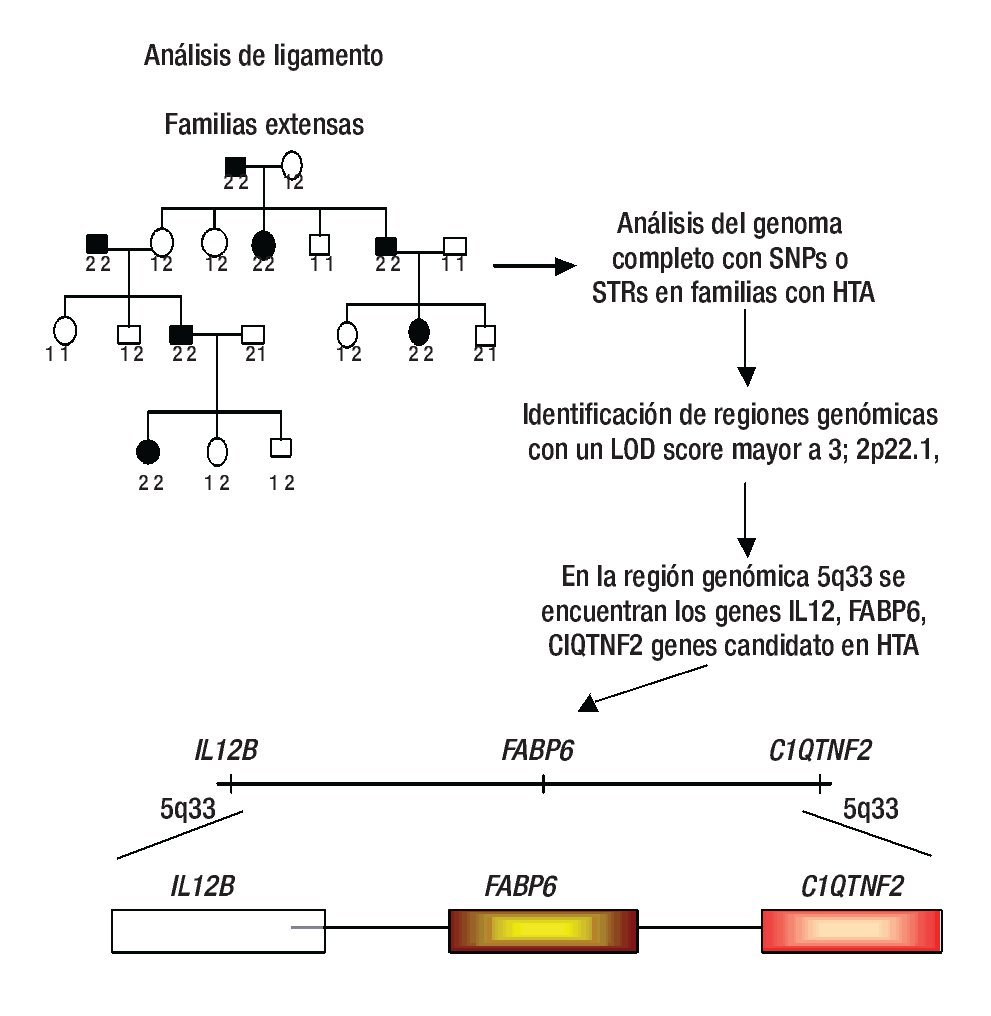
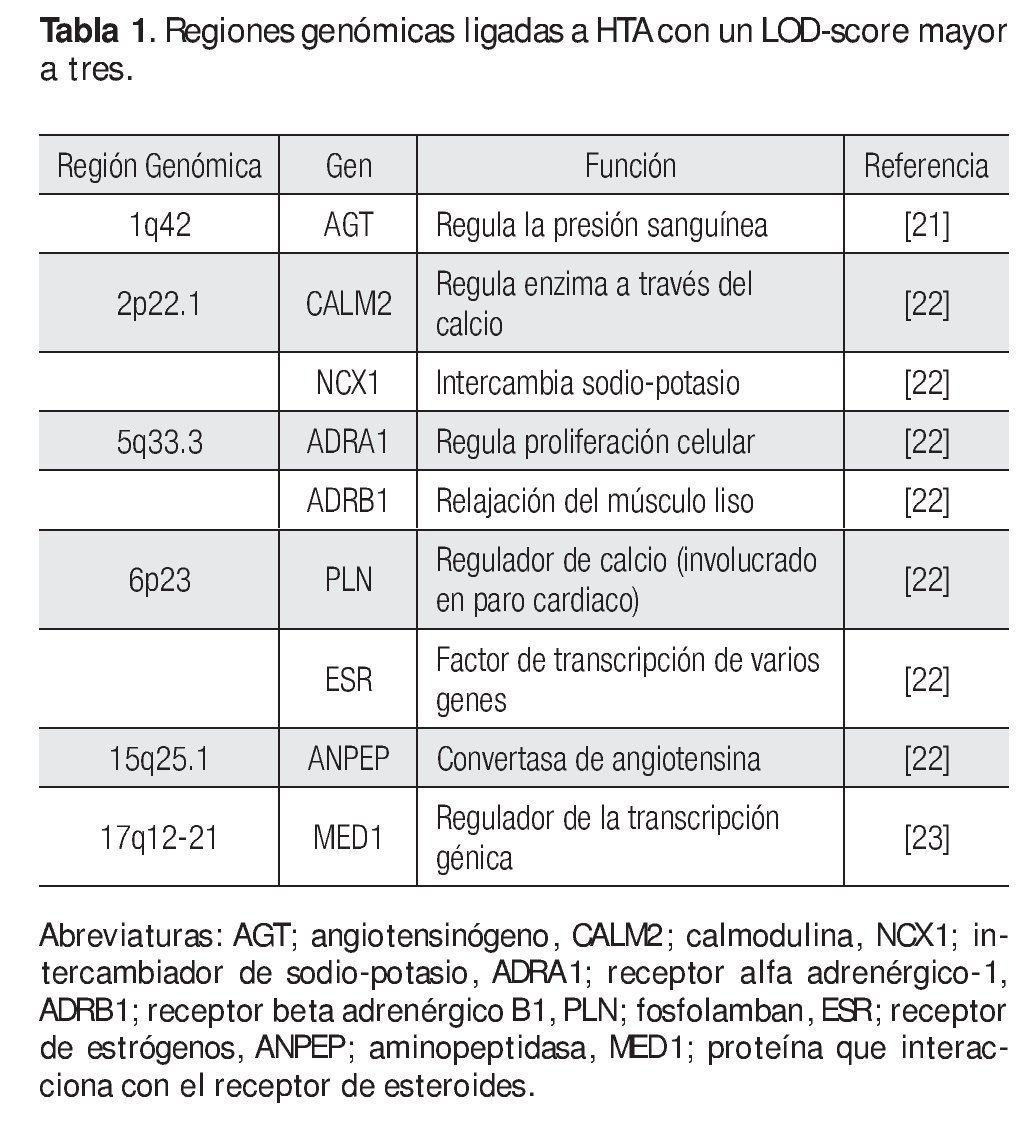
Figura 1. Análisis de ligamiento. En este diseño se emplean a varias familias con múltiples afectados con HTA y la identificación de regiones genómicas se realiza mediante SNPs o STRs. Algunas regiones ligadas a HTA son la 2p22.1, 5q33, entre otras. Los genes IL12B, FASP6 y C1QTNF2 son ejemplos de genes candidato a estudiarse a HTA. Diseño de figura por autores.
Una de las principales ventajas que presentan los análisis de ligamiento en HTA es el análisis con variantes comunes en el genoma completo; esto nos da en primer lugar la identificación de diferentes loci involucrados en HTA en todo el genoma, en segundo lugar, nos sirve para realizar un mapeo genético más detallado con polimorfismos más comunes. Entre las principales desventajas que presenta esta estrategia, está su bajo poder estadístico, debido a la dificultad que implica obtener a un gran número de familias, entre 200 o 300 y con múltiples casos afectados con HTA. Además, la HTA es una enfermedad multifactorial en la que participan varios genes de susceptibilidad de baja penetrancia. A diferencia de las enfermedades monogénicas, en las que un solo gen es el responsable del desarrollo de la patología debido a su alta penetrancia, en las enfermedades multifactoriales hay múltiples genes de baja penetrancia y no hay un patrón de herencia bien definido. Por lo anterior, en las enfermedades complejas como la HTA los estudios de ligamiento han tenido poco éxito para definir cuáles son los genes involucrados en su fisiopatología.19,20
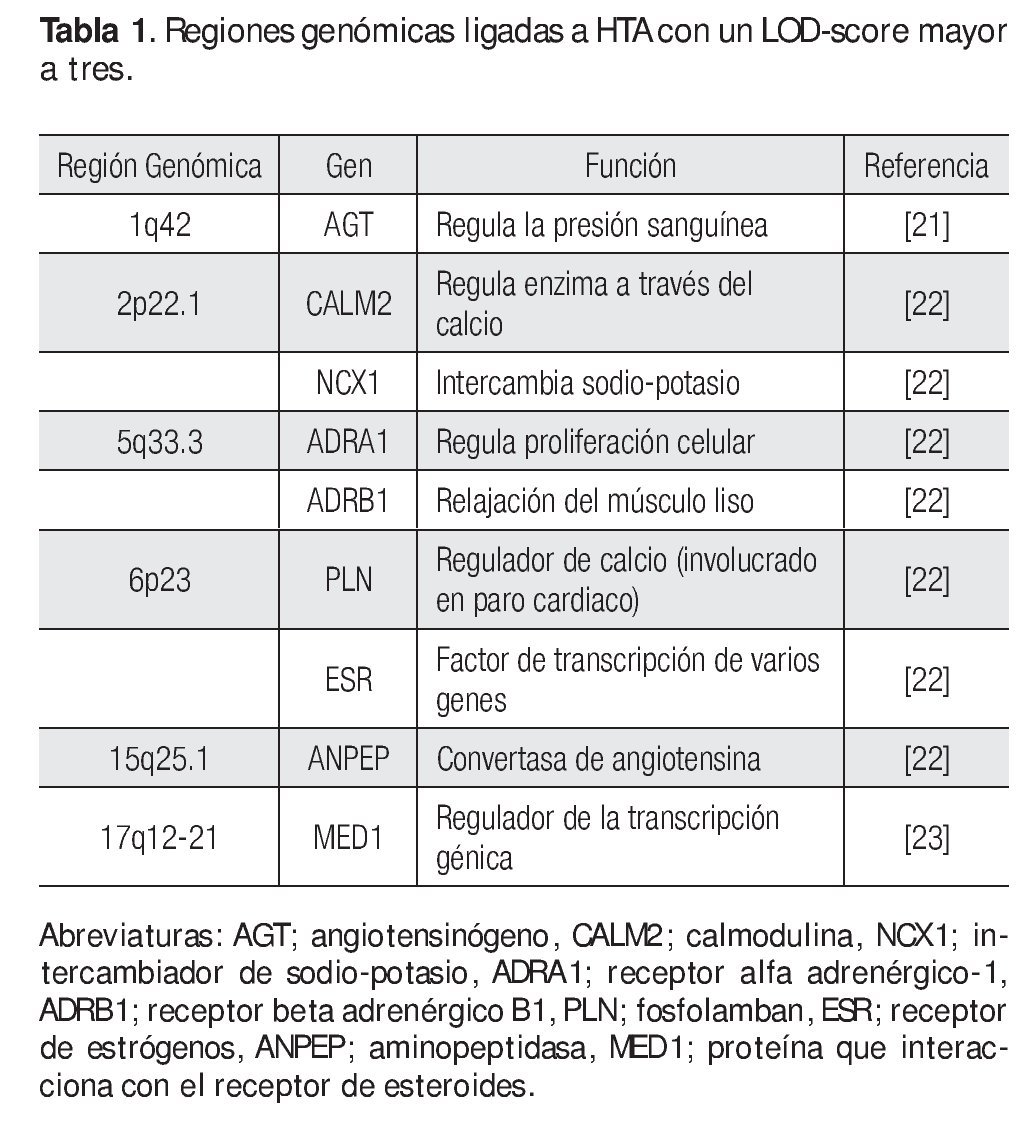
Los GWLS han identificado varias regiones genómicas con un LOD score (logaritmo con base en diez de las probabilidades) mayor a tres (este valor se toma como evidencia de ligamiento y se dice que el locus tiene un ligamiento de 1000 veces a uno respecto a la enfermedad) que están involucradas con HTA, estas incluyen a los siguientes loci: 1q42, 2p22.1, 5q33.3, 6q23, 15q25.1, y 17q12-21 entre otros (Tabla 1).14,21-23 Una vez confirmado el ligamiento, la búsqueda del o de los genes candidato debe iniciarse, en general las regiones ligadas a enfermedades comunes como la HTA son grandes, a veces se tiene que buscar un solo gen en 20 a 30 millones de pb, en esta densidad de nucleótidos comúnmente puede haber más de 500 genes.15 Entonces ¿cómo se pueden buscar genes en una región tan grande? Inicialmente se busca en diversas bases de datos cuales genes están ubicados en la región de ligamiento y cuál de ellos es un buen gen candidato (gen que codifica una proteína que por su función biológica puede estar relacionado con la enfermedad);24 por ejemplo, la primera región de ligamiento involucrada con HTA fue la 1q42, aquí se identificó un marcador río abajo del gen de angiotensinógeno (AGT), que desempeña un papel fundamental en la regulación de la presión arterial a través del sistema renina angiotensina y que se ha asociado con HTA.21 Otra región ligada a HTA es la 5q33.3, en esta región hay una gran cantidad de genes, sin embargo, cuatro de ellos son fuertes candidatos a ser analizados con polimorfismos más comunes como los SNPs, estos genes son IL12B y IL17B (ambos involucrados en eventos inflamatorios), ADAM19 (metaloproteasa involucrada en la adhesión celular y transducción de señales) y FABP6 (la cual tiene un papel importante en la unión a ácidos grasos).
Análisis de asociación
Este tipo de estudio se divide comúnmente en casos y controles, y los basados en familias, en el primero los casos son individuos no relacionados afectados con HTA y controles son individuos sanos no relacionados que no tienen antecedentes de HTA, los segundos, involucran a familias, específicamente tríos: progenitores y afectado.14,15 El objetivo de este diseño de estudio es comparar frecuencias alélicas y genotípicas de la variante estudiada, y entonces observar si existe una diferencia estadística entre los grupos.14,15 Si la variante es más común en los casos que en los controles, decimos que la variante está asociada con susceptibilidad; si es más común en controles que en individuos afectados, entonces confiere protección. En el caso de los estudios de asociación basados en familias, se emplea un método que se denomina análisis de desequilibrio de transmisión (transmission disequilibrium test: TDT), el cual compara la frecuencia con la que los padres heterocigotos transmiten un alelo específico de un marcador genético, por ejemplo tipo SNP al hijo afectado; si la frecuencia del alelo transmitido muestra significancia estadística, se dice que está asociado con la enfermedad.14,15 La gran mayoría de estudios de asociación están realizados en casos y controles, debido a la relativa facilidad que conlleva a reclutar a individuos no relacionados con HTA e individuos sanos sin antecedentes familiares de HTA; sin embargo, para los casos sólo se deben colectar a aquellos que cumplan con criterios clínicos específicos para HTA, mientras que los controles deben ser los adecuados, ya que una gran parte de ellos están mal evaluados, aquí no sólo se debe realizar un cuestionario con historia de antecedentes hereditarios de la enfermedad, deben además existir otros datos clínicos y de laboratorio. Una mala inclusión y estratificación de casos y controles en el estudio puede resultar en falsas asociaciones o descartar a genes que si pueden tener un efecto en la enfermedad. Así, cuando existe evidencia de una región genómica ligada a HTA, lo que sigue es investigar cuáles genes son candidatos a estudiar en esta región mediante un diseño de casos y controles. La elección, de uno o varios SNPs en el gen candidato se pueden hacer de acuerdo a su funcionalidad de la variante dentro del gen.
Actualmente se han desarrollado nuevas metodologías de asociación, denominadas estudios de asociación amplios del genoma o estudio de asociación del genoma completo (GWAS).25,26 Mediante microarreglos de Affymetrix o IIIumina se pueden analizar hasta más de un millón de SNPs;25-27 el parámetro principal es el desequilibrio de ligamiento (linkage disequilibrium: LD), el cual se refiere a una asociación no azarosa de alelos en dos o más loci con la enfermedad.25,28 Estos mismos micro-arreglos permiten además analizar a otro tipo de polimorfismos en el genoma denominados variantes en el número de copias (copy number variants: CNVs), los cuales son segmentos de DNA que tienen un tamaño igual o mayor a 1 kb e incluyen inserciones, deleciones o duplicaciones.27,29,30 Recientemente, los estudios realizados por Tuzun y colaboradores, identificaron 241 CNVs cuando compararon dos genomas de humano.31 Este tipo de variantes no han dado los resultados esperados debido a el tamaño de sondas que identifican a los CNVs.29,32 Sólo algunos CNVs han sido asociados a glomerulonefritis y artritis reumatoide. Sin embargo, estos CNVs han sido detectados mediante gen candidato y no por GWAS.33,34 Hasta ahora, no se han señalado CNVs asociados a HTA .
Causas de asociación de los SNPs
Los estudios de asociación pueden presentar un efecto directo (por su localización dentro del gen puede tener un efecto en el fenotipo de la enfermedad) o indirecto (desequilibrio de ligamiento o estratificación poblacional) entre el SNP analizado y la HTA.
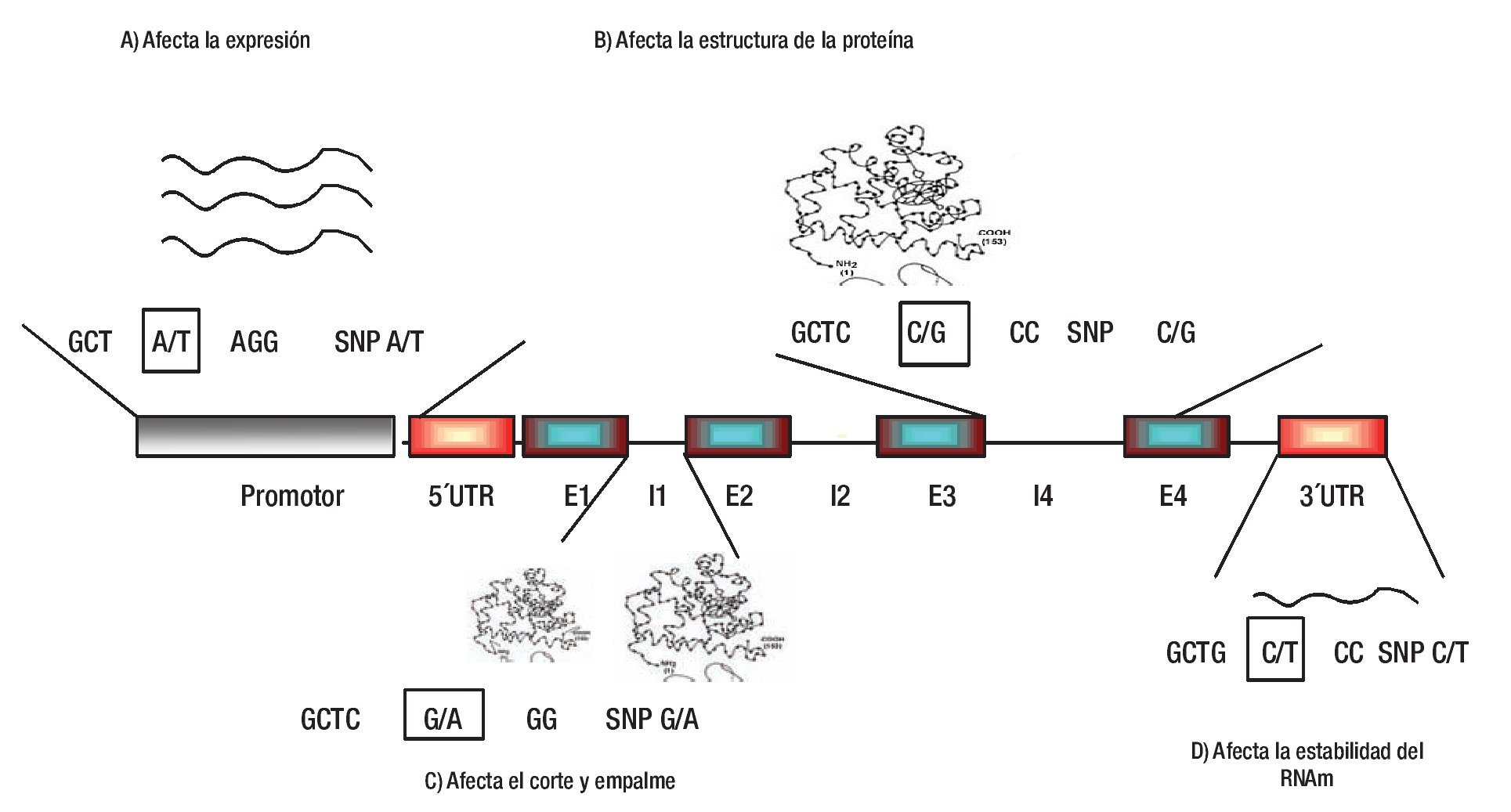
1. Efecto directo del SNP. El SNP puede ser el involucrado directo en conferir susceptibilidad o protección a HTA, debido a que puede tener un papel biológico fundamental en la regulación del gen, síntesis de la proteína, regulación de la maduración, por corte y empalme o en la estabilidad del RNAm (Figura 2).35-39
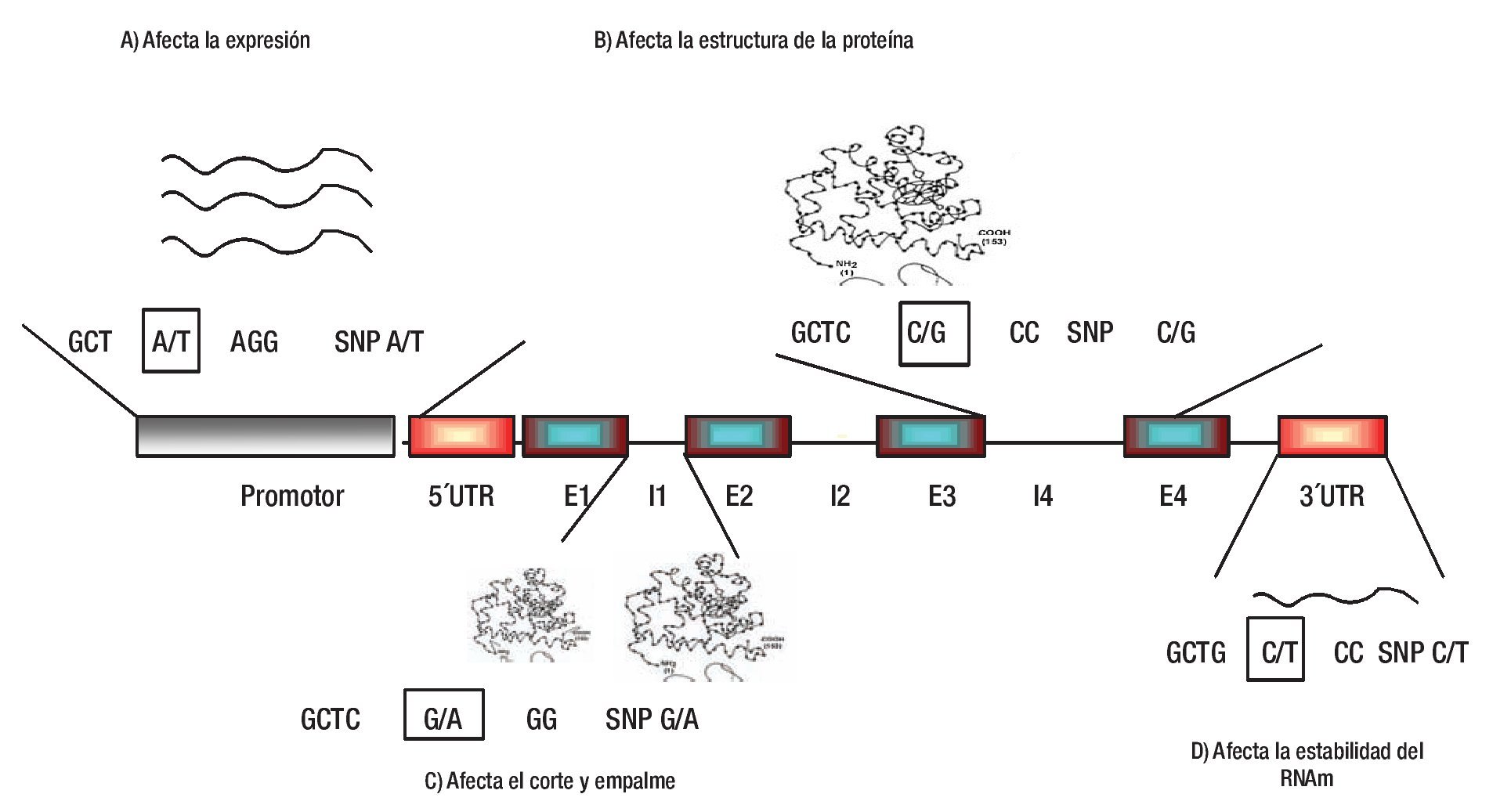
Figura 2. Efecto funcional de los SNPs. Los SNPs se dividen en regulatorios y codificantes (SNPs sinónimos; no cambian aminoácidos, y SNPs no sinónimos; cambian aminoácidos). Los SNPs regulatorios pueden afectar los niveles de expresión, el corte y empalme, la traducción y estabilidad del mensajero (A, C y D). Pocos SNPs codificantes sinónimos pueden afectar a un fenotipo, mientras que los SNPs no sinónimos pueden afectar la actividad y función de la proteína (B). UTR; región no traducida, E; Exón, I; Intrón. Diseño de la figura por autores.
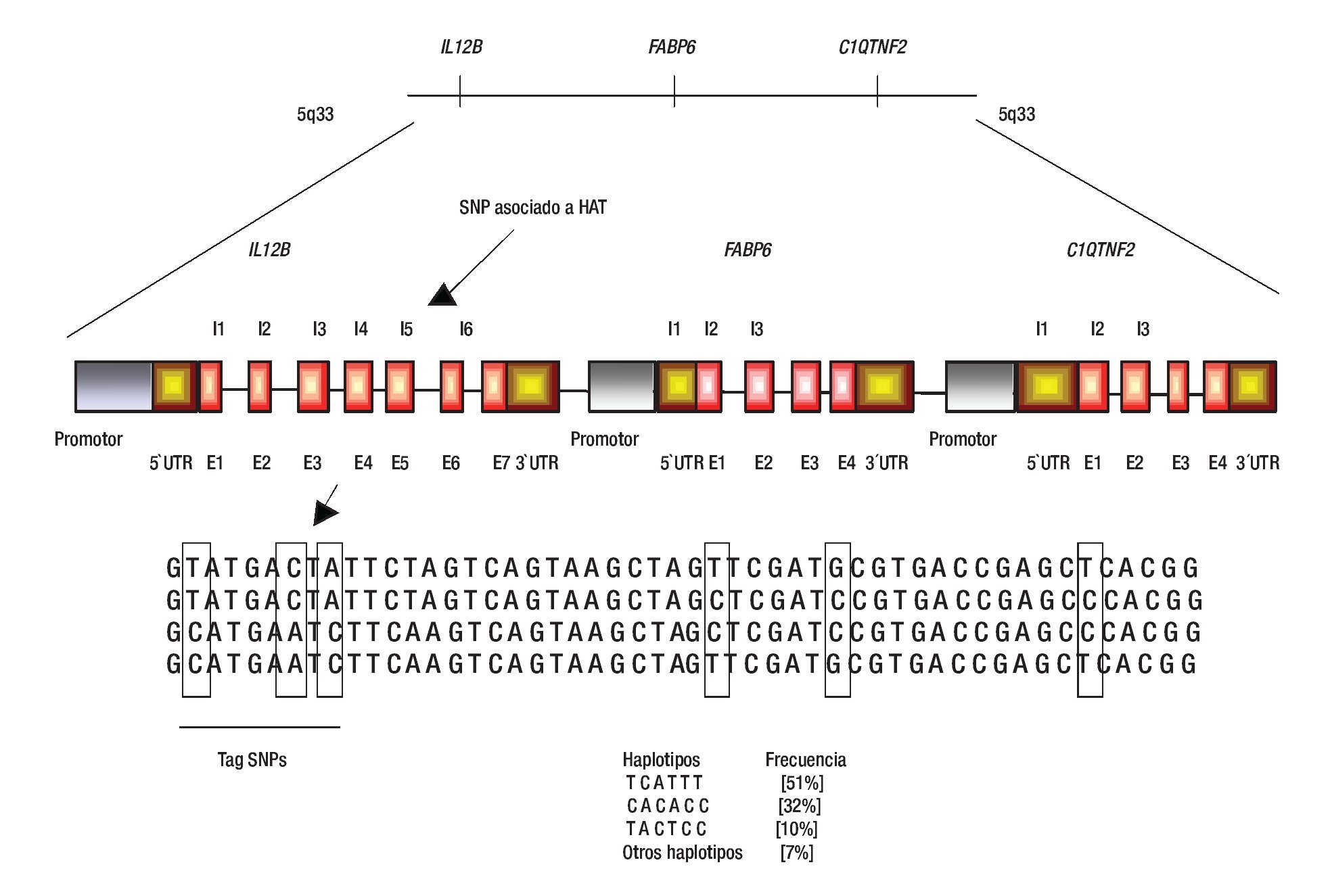
2. Desequilibrio de ligamiento. Algunas veces se asocia un alelo con la enfermedad; sin embargo, es usual que este alelo no sea uno de los involucrados en susceptibilidad o protección sino que una variante cercana o lejana ubicada en el mismo cromosoma sea la verdadera variante involucrada con la susceptibilidad (Figura 3). Este tipo de asociación se debe a LD y se puede presentar tanto en estudios de casos y controles como en los basados en familias.
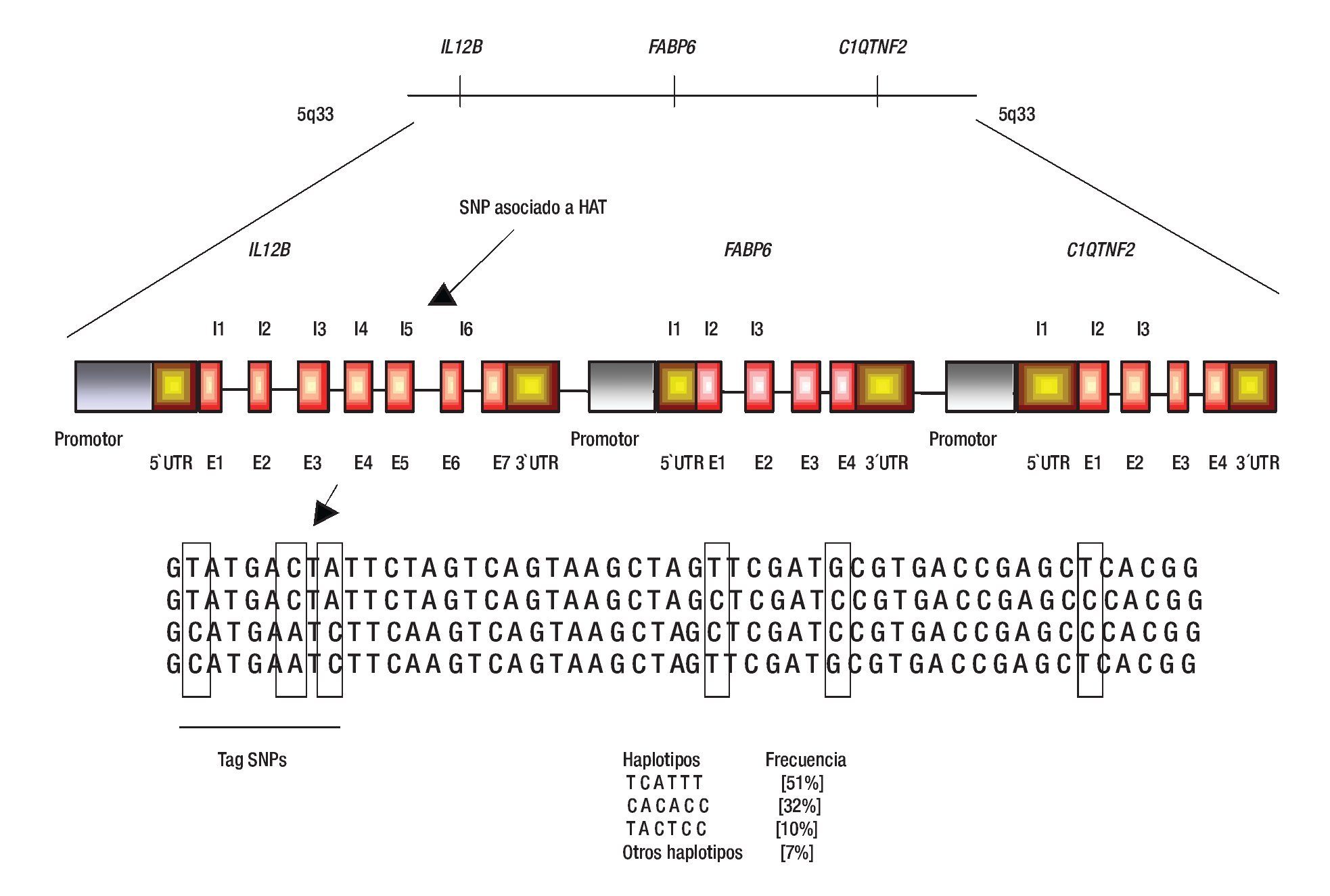
Figura 3. Efecto funcional de los SNPs. Los SNPs se dividen en regulatorios y codificantes (SNPs sinónimos; no cambian aminoácidos, y SNPs no sinónimos; cambian aminoácidos). Los SNPs regulatorios pueden afectar los niveles de expresión, el corte y empalme, la traducción y estabilidad del mensajero (A, C y D). Pocos SNPs codificantes sinónimos pueden afectar a un fenotipo, mientras que los SNPs no sinónimos pueden afectar la actividad y función de la proteína (B). UTR; región no traducida, E; Exón, I; Intrón. Diseño de la figura por autores.
3. Falsas asociaciones debido a estratificación poblacional. En ocasiones, las asociaciones encontradas entre un marcador genético y la enfermedad se deben a estratificación poblacional (sub-grupos de individuos con carga ancestral europea, amerindia o africana y que se presentan en diferente proporción en casos y controles, esto ocasiona en algunas veces que las asociaciones encontradas no sean reales). Mediante el análisis de un grupo de marcadores informativos de historia ancestral en casos y controles, se evita este fenómeno (Figura 4).

Figura 4. Asociación por estratificación poblacional. Si se comparan las frecuencias alélicas y genotípicas del SNP-217 del gen AGT en los diferentes subgrupos poblacionales que representan a los casos y controles habrá una asociación, debido a que el principal subgrupo que representa a los casos tiene el genotipo G/A (europeos), mientras que el que representa al grupo de los sanos es G/G (americanos). Esta asociación se debe a estratificación poblacional; es decir, un subgrupo con un genotipo determinado está sobre-representado en los casos, es necesario hacer un análisis con AIMs para eliminar esta falsa asociación. Diseño de figura por autores.
Estudios de asociación en HTA en genes candidatos: ejemplo del sistema de regulación de la presión sanguínea renina-angiotensina
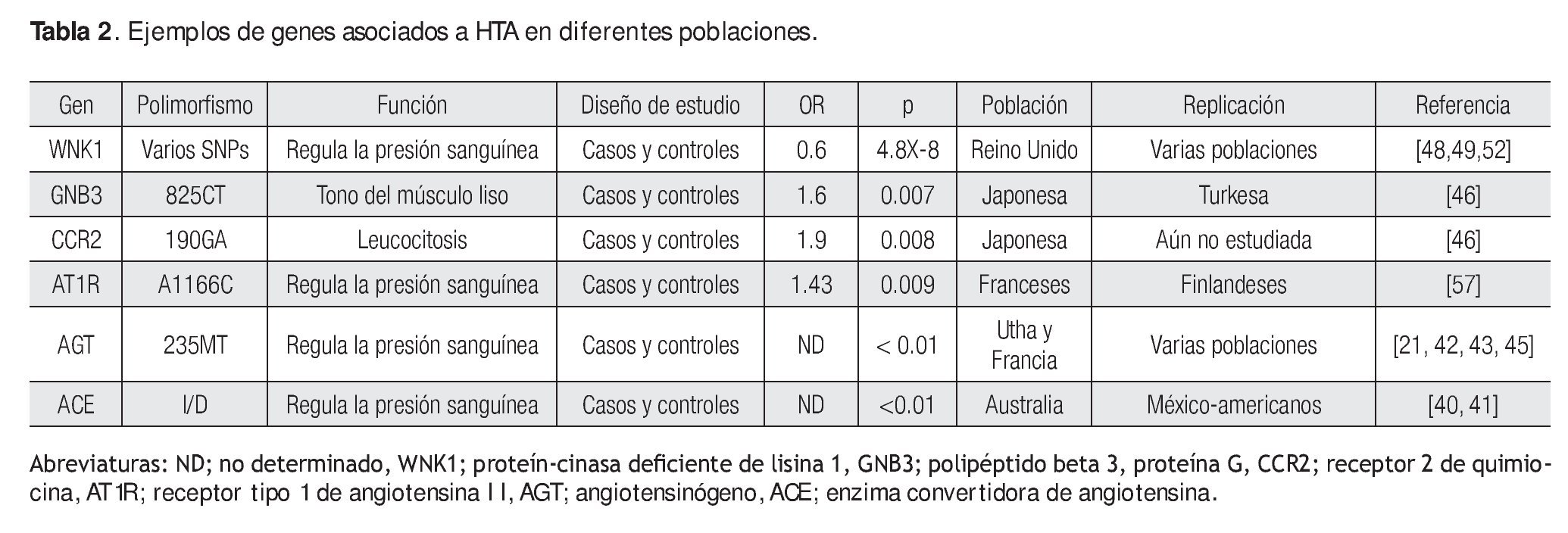
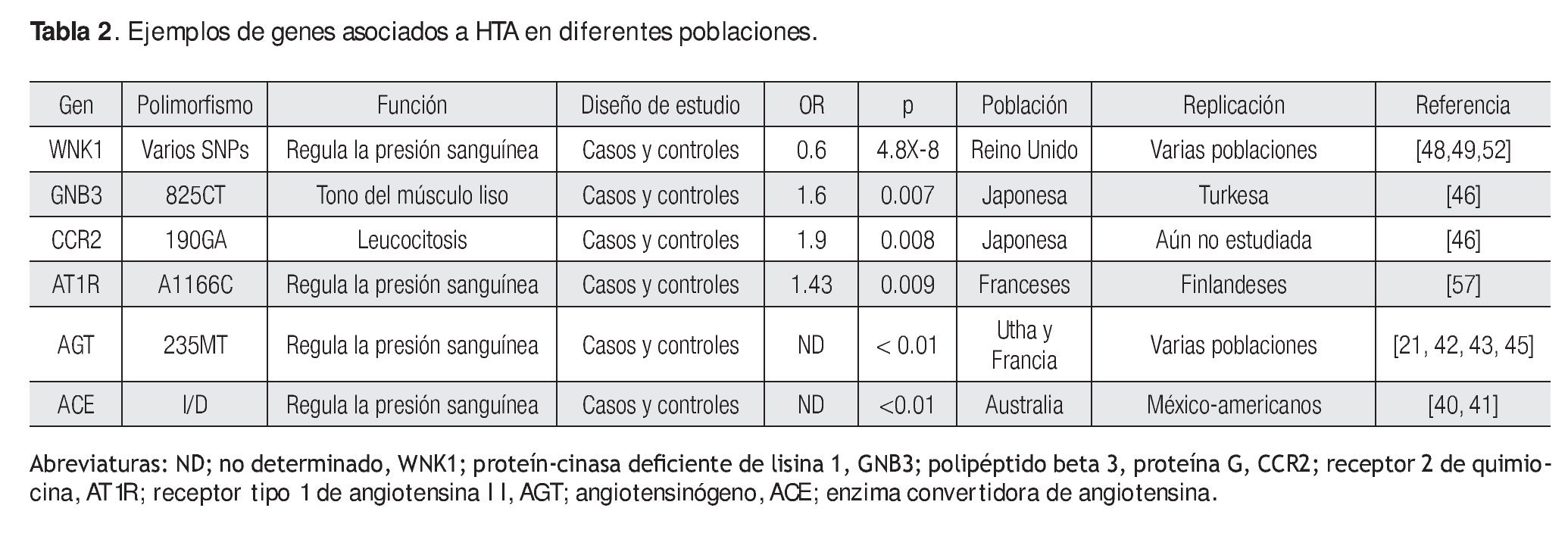
Hasta 2009, los informes sobre SNPs en genes candidatos analizados en HTA fueron pocos.40-53 Sin embargo, a mediados de 2010, la cantidad de publicaciones en HTA u otras enfermedades las que se manifiesta HTA, se ha incrementado drásticamente.54-68 Algunos genes que son excelentes candidatos, porque codifican a proteínas que regulan la presión sanguínea y que participan en el sistema reninaangiotensina, son precisamente la enzima convertidora de angiotensina (Angitiotensin-converting enzime: ACE), angiotensinógeno (angiotensinogen; AGT) y el receptor 1 de angiotensina II (angiotensin II type 1 receptor).14,21,40-49 Aunque existe una gran cantidad de estudios en estos genes, es importante mencionar que no han sido replicados en las diferentes poblaciones de estudio, debido a un sin número de variables, que incluyen tamaño de muestra pequeña, origen étnico, estratificación poblacional, etcétera (Tabla 2).41-43,45-47,57,64,66
De hecho, en tres meta-análisis se evidenció que el genotipo 235T/T del gen AGT confiere susceptibilidad a HTA, un último análisis mostró que esta misma variante confiere protección a HTA. Estos resultados controversiales muestran que estos polimorfismos aún deben de estudiarse en otras poblaciones.42,43,45,47 Una publicación reciente documentó variantes diferentes a la M235T en el gen AGT que muestran asociación con HTA, de hecho un meta-análisis demuestra que la variante T174M y dos variantes ubicadas en la región promotora G-217A y A-20C se asociaron con riesgo a HTA.51 De esta manera, parece que la combinación de los diferentes alelos puede explicar el riesgo a desarrollar HTA, más que sea debido a un sólo SNP en este gen.
Otros genes asociados a HTA
Una publicación reciente, con un tamaño de muestra de 1700 casos y 1700 controles, mostró que un tag SNP (SNP-etiqueta) en el gen WNK1 está asociado con riesgo a presión sanguínea elevada; este mismo resultado fue apoyado cuando se amplió la muestra a 14 451 pertenecientes a seis diferentes poblaciones (Tabla 2); éste y otros informes apoyan la evidencia de que este gen es sumamente importante en el desarrollo de la HTA, de hecho cuando el mismo sufre de algunas mutaciones, causa pseudo-hipoaldosteronismo, una rara enfermedad mendeliana que se caracteriza por presentar HTA e hipercalemia.48,49 Otros genes que se han asociado con HTA son los que codifican para el óxido nítrico sintasa endotelial (endothelial nitric oxide synthase; eNOS), factor de necrosis tumoral-alfa (tumour necrosis factor-alpha; TNF-α), interleucina-6 (interleukine-6; IL-6), entre otros;53,57,64-66 por ejemplo, se ha observado que un SNP ubicado en la subunidad β3 de la proteína G (825C/T) y uno en el gen que codifica al receptor 2 de quimiocinas (190G/A) han sido asociados con susceptibilidad en varones en población Japonesa, mientras que la variante -238G/A de TNF-α se ha asociado en mujeres.46
Estudios de asociación del genoma completo en HTA
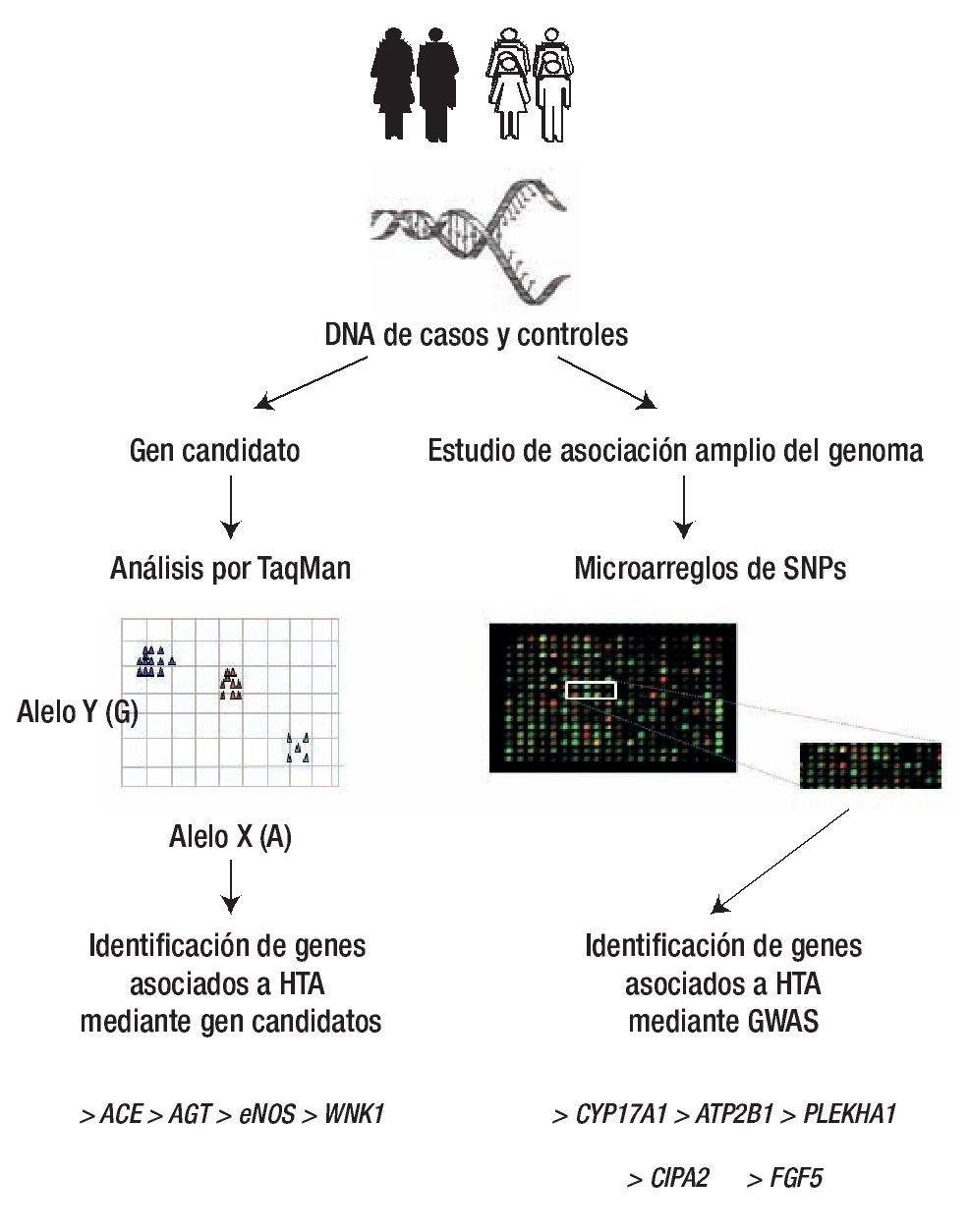
Actualmente, en países desarrollados se han implementado los GWAS mediante micro arreglos de SNPs, estos ensayos sirven para identificar genes asociados a HTA a través del estudio de variantes comunes distribuidas en todo el genoma. Los primeros dos GWAS fueron realizados en 2007 por The Framingham Heart Study (FHS) y por el Wellcome Trust Case Control Consortium (WTCCC); sin embargo, la ausencia de asociaciones significativas (los valores p no fueron estadísticamente significativos; p <5x10-8, valor mínimo necesario para realizar múltiples correcciones) en estos estudios, ocasionó grandes cuestionamientos acerca de la utilidad de esta metodología en la HTA.69-71 El principal problema presentado en el primer estudio fue la incorrecta selección de los controles, ya que no se tomaron los parámetros adecuados en la evaluación de la presión sanguínea que se encuentra comúnmente en individuos aparentemente sanos, este grupo de controles además fue común para siete enfermedades comunes. En el segundo estudio, el análisis se realizó mediante el micro arreglo 100 K, que tiene poca cobertura en el genoma debido a que el chip incluye aproximadamente 100 000 SNPs y no 300 000 como el que utilizó el WTCCC. Un problema común de ambos estudios, fue la no replicación de asociación del gen WNK1, en el cual previamente se había identificado que una sola mutación producía pseudo-hipoaldosteronismo.48,49,52 El problema radicó en que ambos micro arreglos carecieron de Tag SNPs que cubrieran la región del gen WNK1, además, otro problema que presentan los GWAS es la no detección de SNPs con una frecuencia mayor a 5%, de manera que no detectan SNP con frecuencias bajas,72 y en algunas ocasiones estas variantes participan en el desarrollo de las enfermedades. A pesar de los resultados nada alentadores, en el primer estudio algunos SNPs ubicados en los genes ADAMTSL3, CDH13, CCL20, WDR69 mostraron un valor cercano a la asociación con presión sistólica y diastólica. Mientras, que en el segundo GWAS no se identificaron SNPs en genes previamente asociados con HTA y tampoco se identificaron nuevos SNPs asociados con esta patología.11,69-72 Otros 10 GWAS posteriores, no mostraron asociación entre WNK1 y HTA, pero sí identificaron SNPs asociados en los siguientes genes: CDH13, STK39, LOC344371, RASGRP3, IMPG1, PMS1, SLC24A4, YWHA7, IPO7, entre otros (Figura 5).73-82
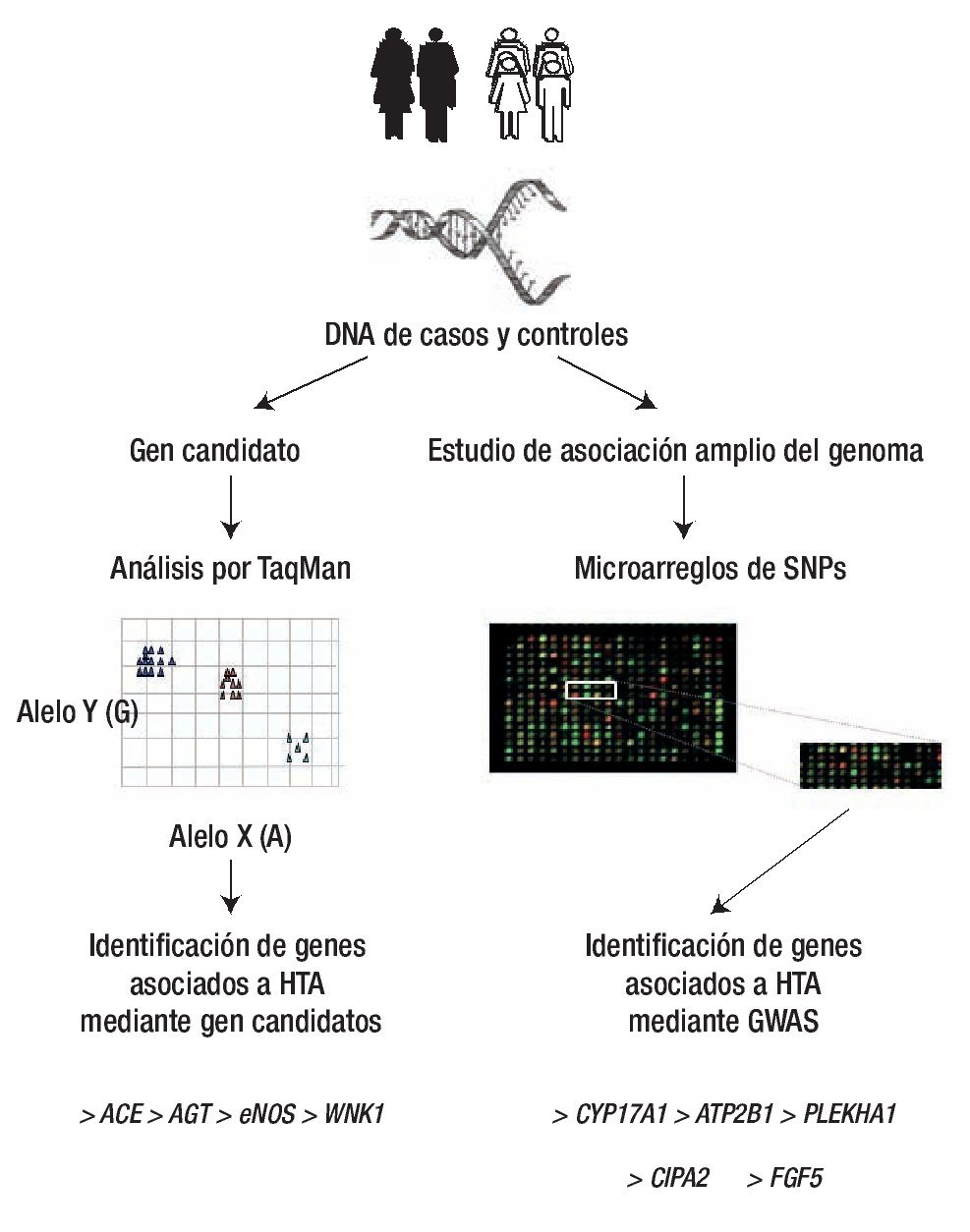
Figura 5. Análisis de asociación casos y controles. La identificación de genes asociación a HTA (ACE, AGT, eNOS, WNK1, entre otros) se ha realizado principalmente mediante gen candidato y con la técnica de TaqMan, mientras que los GWAS realizados con microarreglos han identificado a genes como CYP17A1 y 2, FGF5, SHB3, entre otros. Diseño de figura por autores.
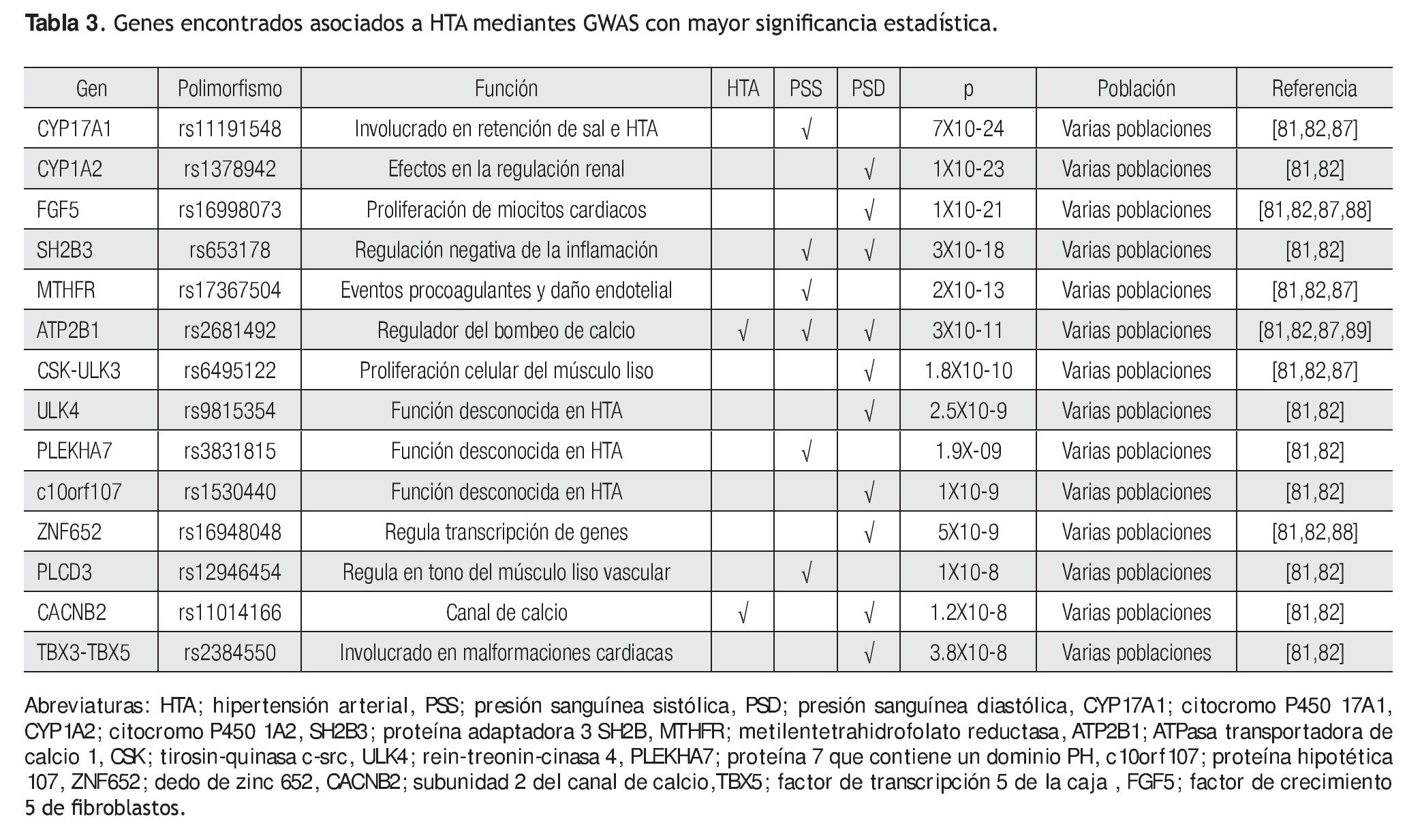
Recientemente, Newton y colaboradores, así como Levy y colaboradores, publicaron dos GWAS con resultados muy robustos, basados principalmente en el gran número de muestras que analizaron (por arriba de 30 mil individuos) y a los valores de p obtenidos (p <5X10-8).81,82 Newton y colaboradores, identificaron a ocho loci asociados con presión sistólica o diastólica que comprenden a los genes que se enlistan en la Tabla 3. Algunos genes fuertemente asociados a HTA son CYP17A1, CYP1A2, FGF5, SH2B3.81 Por otro lado, Ledy y colaboradores identificaron cuatro genes que se asociaron fuertemente a presión sistólica, ATP2B1, CYP17A1, PLEKHA7 y SH2B3 y seis genes asociados con presión diastólica, SH2B3, CSK-ULK3, ULK4, ATP2B1, CACNB2, TBX3-TBX5 y uno para hipertensión, ATP2B1.82
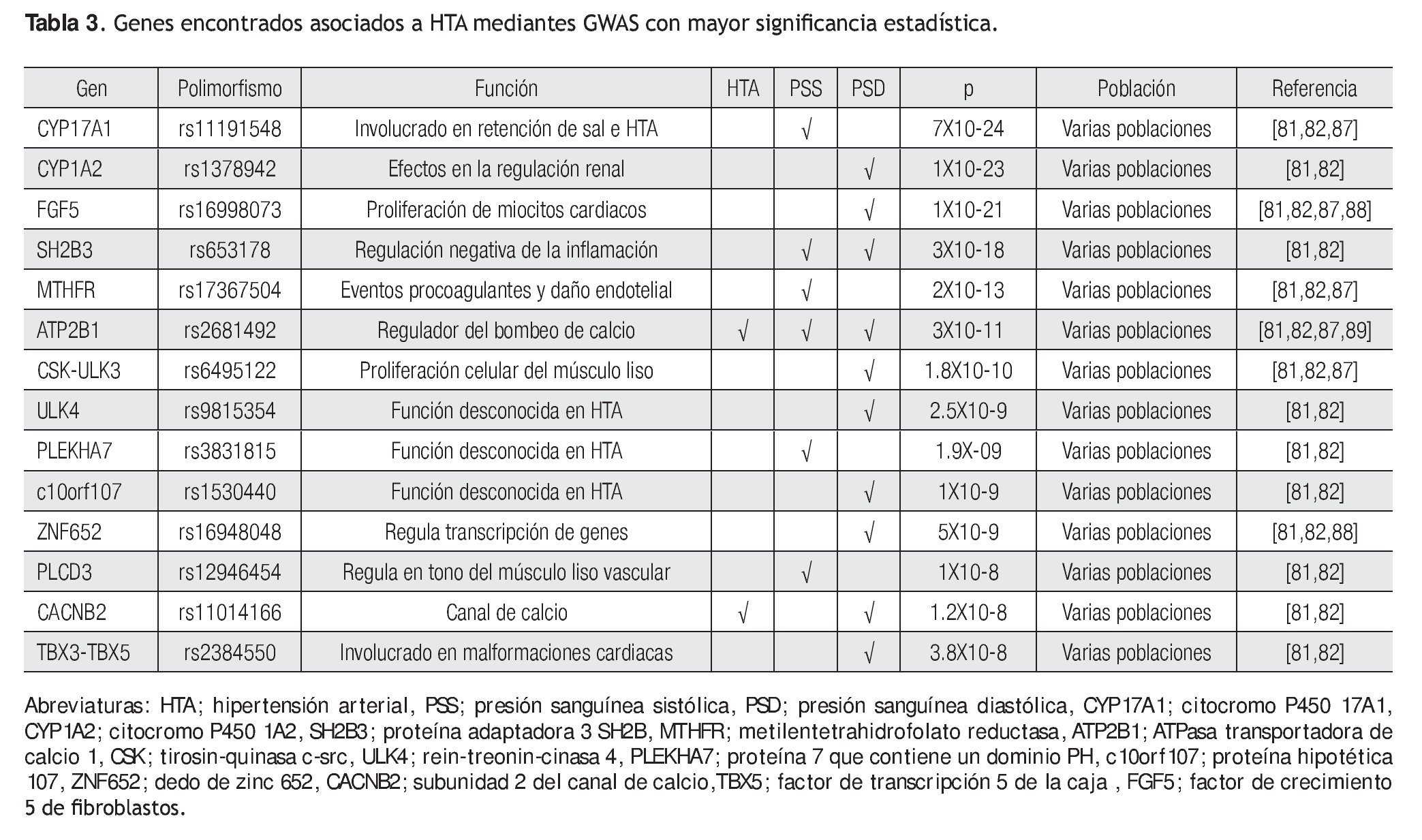
De las variantes encontradas asociadas con HTA, sólo dos genes se comparten en ambos estudios, CYP17A1 y SH2B3. El gen CYP17A1 codifica para una enzima del citocromo P450, que puede regular el tono vascular periférico y la presión sanguínea.83 Este gen ha sido asociado con una forma rara de hipertensión que segrega de manera mendeliana. Por lo anterior, los SNPs del mismo se convierten en candidatos para futuros estudios de casos y controles. Por otro lado, el gen SH2B3 codifica para una proteína adaptadora de linfocitos; este SNP que resultó asociado a presión diastólica, se encuentra en el exón tres y cambia al aminoácido arginina por triptófano (R262W) en la proteína. Este gen se expresa en células precursoras hematopoyéticas y en células endoteliales.84 Ratones knockout para SH2B3 muestran disminución en los progenitores hematopoyéticos, sugiriendo que el alelo de menor frecuencia (Minor allele frequency: MAF) resulte en la pérdida de la función de SH2B3.85 Se ha observado que en respuesta a estímulos inflamatorios, SH2B3 regula negativamente la transducción de señales en células endoteliales, un tipo celular que juega un papel fundamental en la regulación de la presión sanguínea y aterosclerosis. Un tercer gen que se asoció fuertemente a esta patología fue FGF5, este gen codifica a una proteína involucrada en la proliferación y crecimiento de diversos tipos celulares incluyendo a cardiomiocitos, éste además se ha asociado a angiogénesis.86 Cabe aclarar que estos genes identificados mediante GWAS han sido replicados en diversas poblaciones.87-89
Estudios genéticos en enfermedades cardiovasculares en México
En nuestro país, no hay datos de que señalen estudios genéticos y genómicos en esta enfermedad tan prevalente y de consecuencias mortales. Sin embargo, algunos estudios realizados en el Instituto Nacional de Cardiología han arrojado una serie de datos de asociación entre polimorfismos y diversas enfermedades cardiovasculares que incluyen a la enfermedad arterial coronaria, cardiopatía dilatada idiopática, cardiopatía reumática inactiva. Algunos genes analizados y que han mostrado que juegan un papel importante en el desarrollo de estas patologías son HLA-DR, HLA-DQ, TNF-α, entre otros.90 Por otro lado, la variante del tipo inserción/deleción en el gen ACE se asoció a presión sanguínea sistólica91 en mexicoamericanos.
Por otro lado, se analizaron tres polimorfismos que incluyen al -786T/C, Glu298Asp y un repetido en tándem de número variable (Variable number tandem repeat: VNTRs) de 27 pb ubicado en el intrón 4 del gen óxido nítrico sintasa endotelial, aunque el análisis no reflejo una asociación con HTA o presión sanguínea sistólica o diastólica, las variantes -786T/C y el VNTR se asociaron con niveles de triglicéridos y albúmina-creatinina, respectivamente.92 Uno de estos polimorfismos, el Glu298Asp fue también analizado en población mexicana con infarto del miocardio (IAM) con elevación ST, y se encontró una asociación entre el polimorfismo y la enfermedad.93 Por otro lado, se analizó el SNP 677C/T del gen MTHFR que codifica a la 5-10 metilen-tetrahidrofolato reductasa en individuos con IAM con elevación ST y no se encontró asociación entre casos y controles.94 Por otra parte, Meaney (datos no publicados) investigaron un polimorfismo inserción/deleción ubicada en el intrón 16 del gen ACE en THA sin encontrar ninguna asociación entre el polimorfismo y la enfermedad.95
Perspectivas en el tratamiento clínico y la funcionalidad biológica de los SNPs en HTA
Los GWAS y de genes candidato en HTA han dado evidencia acerca de polimorfismos asociados a esta patología; sin embargo, es necesario realizar estudios de replicación en las diferentes poblaciones, incluyendo a la nuestra para saber si éstos causan susceptibilidad. Es bien conocido que entre las poblaciones existe una gran heterogeneidad genética, esto trae la posibilidad que SNPs asociados en esta patología en poblaciones caucásicas no se repliquen en hispanas, africanas u otras. Se debe además evaluar la correlación entre genotipos y el aspecto clínico y de tratamiento, por ejemplo, comúnmente la inserción/deleción ubicado en el intrón 15 de ACE se ha asociado a HTA en diversas poblaciones, algunos fármacos han sido evaluados tomando en cuenta a este polimorfismo; sin embargo, los resultados no correlacionan entre variante y respuesta al tratamiento.96 Entonces, es posible que estas variantes asociadas estén en LD con otra variante que causa susceptibilidad y que aún no han sido identificadas. Recientemente, en este mismo gen ACE se analizó el efecto de tres polimorfismos ubicados en el promotor y se observó que estos alteran los niveles de expresión, esto correlacionó con respuesta al tratamiento a inhibidores de ACE y β-bloquedores.97 Por otro lado, es necesario evaluar el efecto funcional de los SNPs ubicados en los intrones, exones, promotores, 5´ y 3´ UTRs, enhancer, etcétera, en los diversos genes asociados a esta patología, de esta manera podremos determinar la relevancia biológica dentro de una célula, tejido u organismo entero y ver su efecto en el fenotipo hipertenso.
Conclusión
A pesar de que en las últimas décadas se han identificado variantes genéticas asociadas con la HTA, estos estudios han sido poco replicados en las diferentes poblaciones debido a diversos problemas, que incluyen el planteamiento del problema, tamaño de muestra, bajo poder estadístico, estrategia de estudio, estructura de población, etc. Aún así se ha hecho un gran esfuerzo para identificar las variantes genéticas que están implicadas en la etiología de la HTA y de las enfermedades cardiovasculares. Recientemente se ha desarrollado una estrategia nueva que incluye estudiar ampliamente o completamente el genoma mediante micro arreglos de SNPs. Desafortunadamente este tipo de tecnología no se ha aplicado en la mayoría de países en vías de desarrollo debido a su elevado costo y al número de muestras que se requieren para obtener datos contundentes. Es importante hacer notar que los estudios de GWAS son relativamente nuevos en las enfermedades multifactoriales, donde se incluye a la HTA. Sin embargo, este tipo de análisis nos muestra un panorama general acerca de las variantes en el genoma asociadas con ciertas características, como la presión sistólica o diastólica altas o HTA. Es importante mencionar que aunque parece que estamos en una etapa donde estos estudios de GWAS parecen resolver todo, no es así debido a que también presentan varios inconvenientes como bajo desequilibrio de ligamiento en varias regiones del genoma, además de no capturar SNPs con una frecuencia menor a 5%, y en algunas ocasiones estas variantes sí participan en el desarrollo de la enfermedad. Así, la completa identificación de los factores genéticos de riesgo en HTA tendrá en un futuro no muy lejano un gran potencial en el desarrollo de nuevos métodos de diagnóstico, tratamiento y predicción de la enfermedad.
Correspondencia: José Manuel Fragoso.
Juan Badiano N°1, Tlalpan 14080, México D. F.
Teléfono: (525) 5573 2911, ext: 1460. Fax: (525) 5573 0926.
Correo electrónico: mfragoso1275@yahoo.com.mx.
Recibido el 24 de septiembre de 2010;
aceptado el 31 de marzo de 2011.