


Atrial fibrillation (AF) is the most common sustained chronic cardiac arrhythmia in clinical practice, which increases the risk of stroke and thromboembolism and is an independent predictor of mortality. The underlying mechanisms involved in the development of AF have yet to be fully elucidated. However, once initiated, AF tends to self-perpetuate, owing to structural and electrical remodeling in the atria.
MicroRNAs (miRNAs) represent a sizable sub-group of small non-coding RNAs, which degrades or inhibits the translation of their target mRNAs, thus regulating gene expression and playing an important role in a wide range of biologic processes. Clinically, there is increasing evidence of the potential diagnostic role of miRNAs as biomarkers, representing a novel therapeutic target in AF.
The aim of this review is to provide an exhaustive overview of the role of miRNAs in AF and to discuss the diagnostic and therapeutic potential of miRNAs in this arrhythmia.
La fibrilación auricular (FA) es la arritmia cardíaca sostenida crónica más común en la práctica clínica, lo que aumenta el riesgo de accidente cerebrovascular y tromboembolismo, y es un predictor independiente de mortalidad. Los mecanismos subyacentes implicados en el desarrollo de la FA todavía no se han aclarado completamente. Sin embargo, una vez iniciada, la FA tiende a perpetuarse, debido al remodelado estructural y eléctrico en la aurícula.
Los microARN (miARN) representan un subgrupo importante de pequeños ARN no codificantes, que degradan o inhiben la traducción de sus ARN mensajeros diana, regulando así la expresión génica y que desempeñan un papel importante en una amplia gama de procesos biológicos. Clínicamente, se ha observado con creciente interés el posible papel diagnóstico de los miARN como biomarcadores, representando una nueva diana terapéutica en la FA.
El objetivo de esta revisión es proporcionar una visión exhaustiva de la función de los miARN en la FA y discutir el posible papel diagnóstico y terapéutico de los miARN en esta arritmia.
Atrial fibrillation (AF) is the most common arrhythmia in clinical practice and is associated with decreased quality of life, and increased mortality and morbidity from stroke and thromboembolism.1 The precise mechanisms for the development of AF are not completely understood, but are thought to be multifactorial.2 It is hypothesized that initiation of AF may be due to the effect of a number of pathophysiological processes, including inflammation, oxidative stress and fibrosis. These processes could play an important role in the initiation of AF producing a structural and electrical remodeling of the cardiac tissue that allows the arrhythmia to be maintained.1 Despite the recent advances in pharmacological approaches to prevent the recurrence of AF, none of the currently available therapies are fully effective in the long-term.3 A major reason for the limited success of current therapies may be the fact that they do not address the upstream underlying pathophysiologies of AF and if they do so, only target a single mechanism, whereas AF is a complex, continuous process and changes associated with aging.4 Thus, an improved understanding of the mechanisms underlying AF is essential for the development of novel therapeutic approaches.5
A recent paper published by Orenes-Piñero et al. provide an overview of the impact of several microRNAs (miRNAs) in electrical and structural remodeling of the cardiac tissue, and the diagnostic and therapeutic potential of miRNAs in cardiovascular disease.6 Cardiac remodeling is an adaptive, regulatory process of cardiac myocytes that occurs over time in order to maintain homeostasis against external stresses.7 This may occur at the ionic, genomic, cellular, and extracellular levels.8 If a stressor is persistent, the process can become irreversible and cellular and extracellular changes such as apoptosis, necrosis, and fibrosis can occur.8 Two principal forms of remodeling have been described: (i) structural remodeling, which alters cardiac tissue architecture and (ii) electrical remodeling, which alters cellular electrical properties.9
miRNAs are endogenous, conserved, single stranded, small (approximately 22 nucleotides in length), non-coding RNAs that repress gene expression at the post-transcriptional level by targeting mRNA.10 Since 1993, when the first miRNA was discovered,11 and according to the latest miRBase release (v20, June 2013) more than 24,500 miRNA loci from 206 species, processed to produce 30,424 mature miRNAs have been discovered. Among them, the human genome encodes more than 2600 mature miRNAs, which may target over 60% of human protein-coding genes. Each miRNA regulates several different target genes12 by annealing to complementary sequences in the 3′-untranslated regions (3′UTR) of target mRNAs of protein-coding genes, causing mRNA to be cleaved or repressing the translational machinery needed for protein synthesis.13
Search strategyPublished data for this review were identified by search and selection in MEDLINE database and reference lists from relevant articles and reviews. A two-step approach was used. First, miRNAs were identified in a search with the keywords “AF” and “miRNA”. Second, miRNAs that were identified with this search related with AF were used as keywords with the addition of the following keywords “electrical remodelling”, “structural remodelling”, “fibrosis”, “ion channels” and “oxidative stress”. Bibliographies of all selected articles and review articles about AF and/or miRNAs were reviewed for other relevant articles.
Micro RNAs involved in AFThe most up-to-date research data on the roles of miRNAs in AF initiation and maintenance show that miRNA-induced gene expression changes could contribute to electrical and structural remodeling that perpetuates AF. A comprehensive summary of the most important miRNAs associated to AF initiation and maintenance will be discussed in this manuscript.
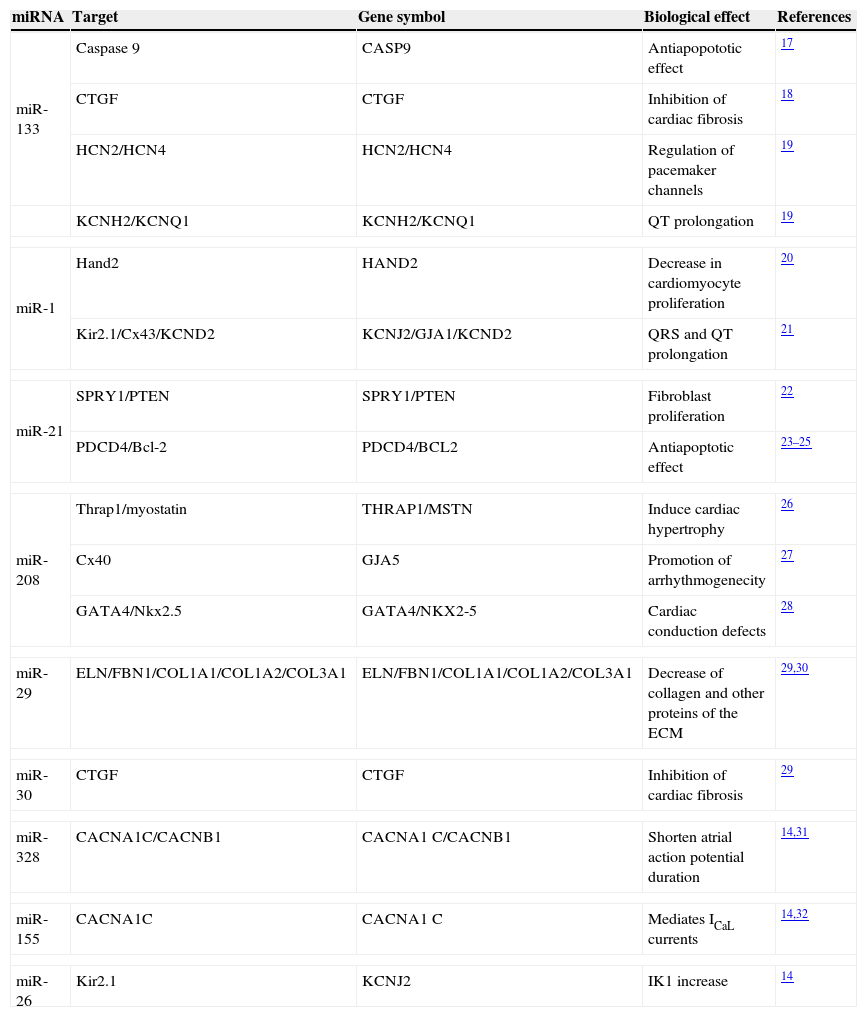
miR-133This miRNA is the most common miRNA in human heart.14 miR-133 is a muscle-specific miRNAs that regulates protein levels by repressing translation of genes involved in cardiac contractility, hypertrophy, and electric conductance.15 TargetScan prediction showed more than 500 putative miR-133 mRNA targets, and numerous functional roles have been proposed, including preventing genetic cardiac hypertrophy15 or suppressing embryonic cardiomyocyte proliferation.16 In addition, miR-133 has shown an anti-apoptotic role. Multiple target sites for miR-133 throughout the sequence of the caspase 9 gene have been identified, thus inhibiting its apoptotic effect17 (Table 1). Furthermore, miR-133 expression decreases fibrosis by connective tissue growth factor (CTGF) downregulation18 and represses HCN2 pacemaker channel and ERG K+ channel expression19 (Table 1). For these reasons, therapeutic overexpression of miR-133 in vivo has been proposed to prevent cardiac remodeling and AF maintenance.
Biological functions regulated by miRNAs affecting AF.
| miRNA | Target | Gene symbol | Biological effect | References |
|---|---|---|---|---|
| miR-133 | Caspase 9 | CASP9 | Antiapopototic effect | 17 |
| CTGF | CTGF | Inhibition of cardiac fibrosis | 18 | |
| HCN2/HCN4 | HCN2/HCN4 | Regulation of pacemaker channels | 19 | |
| KCNH2/KCNQ1 | KCNH2/KCNQ1 | QT prolongation | 19 | |
| miR-1 | Hand2 | HAND2 | Decrease in cardiomyocyte proliferation | 20 |
| Kir2.1/Cx43/KCND2 | KCNJ2/GJA1/KCND2 | QRS and QT prolongation | 21 | |
| miR-21 | SPRY1/PTEN | SPRY1/PTEN | Fibroblast proliferation | 22 |
| PDCD4/Bcl-2 | PDCD4/BCL2 | Antiapoptotic effect | 23–25 | |
| miR-208 | Thrap1/myostatin | THRAP1/MSTN | Induce cardiac hypertrophy | 26 |
| Cx40 | GJA5 | Promotion of arrhythmogenecity | 27 | |
| GATA4/Nkx2.5 | GATA4/NKX2-5 | Cardiac conduction defects | 28 | |
| miR-29 | ELN/FBN1/COL1A1/COL1A2/COL3A1 | ELN/FBN1/COL1A1/COL1A2/COL3A1 | Decrease of collagen and other proteins of the ECM | 29,30 |
| miR-30 | CTGF | CTGF | Inhibition of cardiac fibrosis | 29 |
| miR-328 | CACNA1C/CACNB1 | CACNA1C/CACNB1 | Shorten atrial action potential duration | 14,31 |
| miR-155 | CACNA1C | CACNA1C | Mediates ICaL currents | 14,32 |
| miR-26 | Kir2.1 | KCNJ2 | IK1 increase | 14 |
Abbreviations: CTGF: connective tissue growth factor; HCN2, KCNH2, KCNQ1, KCNJ2, KCND2: potassium ion channels; Hand2: heart and neural crest derivatives-expressed protein 2; Cx: connexin; SPRY: sprouty homolog; PTEN: phosphatase and tensin homolog; PDCD4: recombinant human programmed cell death 4Bcl-2: B-cell CLL/lymphoma 2; Thrap1: thyroid hormone receptor associated protein 1; Nkx2.5: NK2 homeobox 5; ELN: elastin; FBN1: fibrillin-1; COL: collagen; CACN: calcium channel.
miR-1 is the second more abundant miRNA in humans, with around 1/3 of the miR-133 levels.14 Like most miRNAs, miR-1 has been predicted to target hundreds of genes, but one validated target of miR-1 in the heart is Hand2, an important cardiac transcription factor whose genetic ablation in mice produced failure in ventricular myocytes.20 The role of miR-1 inhibiting cardiac hypertrophy by several pathways has been clearly demonstrated. In addition, miR-1 has shown to be pro-apoptotic, in contrast to miR-133.17 On the other hand, application of miR-1 into the myocardium induces arrhythmias by targeting KCNJ2 (encoding the Kir2.1 channel subunit) which is the channel responsible for the inward rectifier K+ current (IK1) and Connexin 43 (Cx43) the primary gap junction protein in the ventricle, among other ion channels.21 Together, all these data indicate that alteration in miR-1 levels may be a trigger for the production of the arrhythmogenic substrate.
miR-21miR-21 is one of the most highly upregulated miRNAs during cardiac remodeling. The impact of miR-21 on cardiac fibrosis has been well established. Importantly, blocking of endogenous miR-21 reduced genes encoding collagens and extracellular matrix molecules that are highly upregulated during cardiac fibrosis.22 In addition, miR-21, prevented cell damage in cultured cardiac myocytes subjected to oxidative stress at least in part through targeting PDCD4 protein (Recombinant Human Programmed Cell Death 4 protein).23 It has been classified as an oncomir due to its anti-apoptotic function and as a result of being deregulated in several type of cancers.24 Bcl-2, a pro-apoptotic signal molecule, has been demonstrated to be a target of miR-2125 (Table 1), thus supporting the anti-apoptotic role of this miRNA.
miR-208Cardiac overexpression of miR-208 is sufficient to activate calcineurin signaling, cardiac pressure-overload-induced stress and induces cardiac hypertrophy by repressing the expression of thyroid hormone-associated protein 1 (Thrap1) and myostatin.26 Moreover, the electrocardiograms of approximately 80% of the miR208 knockout mice lack P waves preceding QRS complexes, suggesting that miR208−/− mice suffer from AF. The lack of miR208 results in deficiencies in Cx40 transcript expression, a molecule restricted to the atria and the specialized cardiomyocytes that constitute the His bundle and Purkinje fibers.27 Apart from regulating Cx40 expression, miR208 can directly repress the expression of GATA4 and the Nkx2.5, two transcriptional cofactors expressed within the cardiac conduction system of the adult heart28 (Table 1). All these observations demonstrate the role of miR-208 as an anti-hypertrophic miRNA and highlight its importance in the electric remodeling that perpetuates AF in human heart, thus being a potential target in therapeutic purposes.
miR-29This miRNA is expressed preferentially in fibroblasts. It has been observed that elastin (ELN), fibrillin 1 (FBN1) and several types of collagen, such as COL1A1, COL1A2 and COL3A1, are potential targets for miR-2929 (Table 1). Moreover, in vivo downregulation of miR-29 correlated with an upregulation of these proteins and with an amount of collagen deposition. Remarkably, miR-29, miR-133 and miR-30 are the most strongly fibrosis-associated miRNAs targeting a great number of extracellular-matrix-related mRNAs.29 In a recent study, it was observed that myocardial infarction (MI) treatment with propanolol increased the expression of miR-29 in rats.30 This can be one of the explanations of the reversal effects of this β-adrenoceptor blocker in MI.
Other miRNAs involved in AFThe levels of many miRNAs have been demonstrated to be altered in AF by a series of high-throughput miRNA microarray analysis. miR-30 and miR-328 regulate atrial electric remodeling through targeting L-type Ca2+ channel genes.14,31 Similarly, miR-155 mediates ICaL currents through the calcium channel 1C (CACNA1C).32 Moreover, miR-26 plays a significant role in upregulating IK1 in AF.33 Regarding miR-328, two genes, CACNA1C and CACNB1, which encode cardiac L-type Ca2+ channel α1c- and β1 subunits, respectively, were identified as their potential targets31 (Table 1). In an in vivo study, it was found that knockdown of miR-328 increased L-type Ca2+ current and lengthened atrial action potential duration, being a useful strategy to recover sinus rhythm in AF.31 Inversely, downregulation of miR-26 results in relief of repression of KCNJ2/Kir2.1, and thereby an increase in IK1, leading to the shortening of atrial action potential and creating the substrate for AF.14
miRNA perspectives: potential uses and therapeutic strategiesThe biology of miRNAs is a relatively new research area. In recent years, miRNAs are emerging as pivotal modulators of mammalian cardiovascular development and disease, including hypertrophy, heart failure, arrhythmias, cardiac injury and so on. Each miRNA can regulate several mRNAs with similar function, thus impacting complex biological processes.
To recover the decreased miRNA levels in a particular cardiovascular disease and thus prevent the pathological process, a miRNA re-expression strategy can be used to increase miRNA levels. miRNA Mimic (miR-mimics) are synthesized nucleic acids that bind to target mRNAs similarly as the natural miRNAs, having the same post-transcriptional regulatory effects and repressing the target gen to be transcripted.19 Inversely, for those miRNAs that are upregulated and cause a disease, the anti-miRNA techniques to block miRNA expression can be employed. One of them is the use of antisense inhibitor oligonucleotides (AMOs). AMOs are non-naturally designed oligonucleotides that are fully complementary and bind to their target miRNAs reducing their activity. The success of both techniques have been already demonstrated.19,34
Future studies aimed at understanding how miRNAs are integrated into heart disease are a prerequisite for their development as potential therapeutic targets. Significantly, a number of reports here reviewed have demonstrated the feasibility of these approaches in pre-clinical studies. However there is still a long way to go for miRNA-based therapeutic strategies in human. Effective delivery of specific miRNAs (natural or synthetic) to the specific targets (e.g., organs, tissues, or cell types) is the real major challenge, although in theory, miRNAs can be a very powerful weapon in our hands to subjugate AF.
FundingNo endorsement of any kind received to conduct this study/article.
Conflict of interestThe authors declare no conflict of interest.