


Se expone el caso de una niña de 5 años de edad que presenta un cuadro malformativo compatible con síndrome de Rubinstein-Taybi (RTS). Se la remite al cardiólogo para valoración por soplo sistólico sin otra sintomatología. A la exploración física se encontraba: talla en percentil 5, fenotipo con pulgares gruesos así como soplo holosistólico en mesocardio ll-Vl, pulsos normales. FISH positivo para deleción del cromosoma 16p13.3. En el estudio ecocardiográfico se detectó una comunicación interventricular perimembranosa pequeña con mecanismos de cierre, presión pulmonar normal, pero en los planos de grandes vasos se visualizaron 2 arcos aórticos bien desarrollados. Tras el diagnóstico de doble arco aórtico y sin que la paciente tuviera ninguna sintomatología respiratoria ni digestiva se realizó un estudio completo con esofagograma que mostraba compresión extrínseca y una fibrobroncoscopia que revelaba una traqueomalacia leve y compresión extrínseca traqueal por doble arco aórtico y lobulación pulmonar anormal. Se realizó un cateterismo cardiaco que confirmó los hallazgos previos.
La niña fue remitida a nuestro centro para tratamiento quirúrgico. Debido a la ausencia de sintomatología se realizó una TAC multicorte que mostró un anillo vascular completo con doble arco aórtico, con ligera asimetría en el calibre de los arcos siendo el izquierdo ligeramente de mayor calibre. (fig. 1). No se observó ninguna compresión significativa de la vía aérea (fig. 2) y dada la ausencia de síntomas no se realizó intervención quirúrgica.
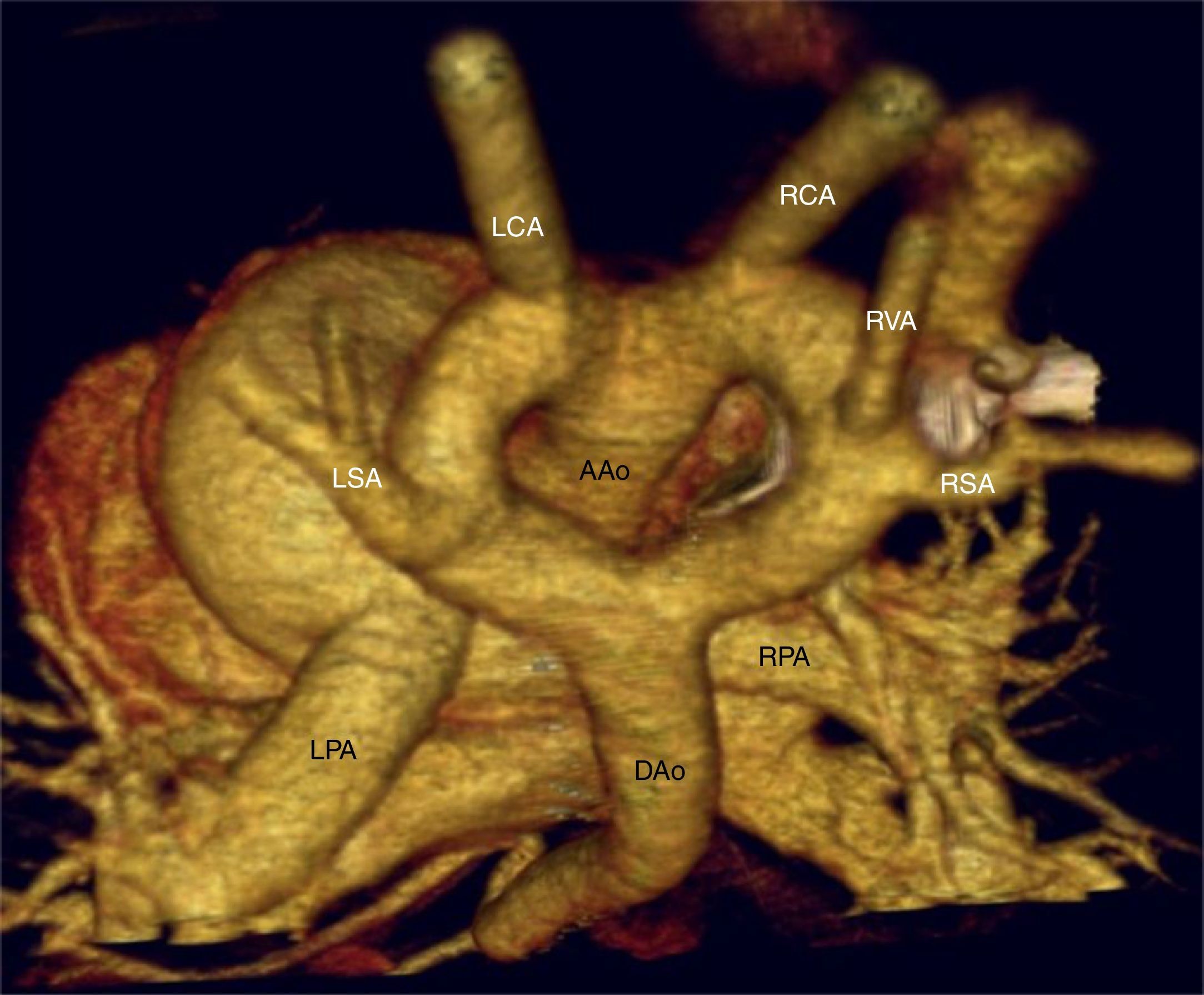
; DAo: descending aorta (aorta descendente); LCA: left carotid artery (arteria carótida izquierda); LPA: left pulmonary artery (arteria pulmonar izquierda); LSA: left subclavian artery (arteria subclavia izquierda); RCA: right carotid artery (arteria carótida derecha); RPA: right pulmonary artery (arteria pulmonar derecha); RSA: right subclavian artery (arteria subclavia derecha); RVA: right vertebral artery (arteria vertebral izquierda).")
Angio-TC torácica con reconstrucción VR-3D, visión posterosuperior. En esta imagen podemos observar un arco aórtico completo, con origen independiente de los troncos supraaórticos: en el arco izquierdo, ligeramente de mayor calibre, se originan la arteria carótida y subclavia izquierdas y en el arco derecho, la arteria carótida, vertebral y subclavia derechas. AAo: ascending aorta (aorta ascendente); DAo: descending aorta (aorta descendente); LCA: left carotid artery (arteria carótida izquierda); LPA: left pulmonary artery (arteria pulmonar izquierda); LSA: left subclavian artery (arteria subclavia izquierda); RCA: right carotid artery (arteria carótida derecha); RPA: right pulmonary artery (arteria pulmonar derecha); RSA: right subclavian artery (arteria subclavia derecha); RVA: right vertebral artery (arteria vertebral izquierda).
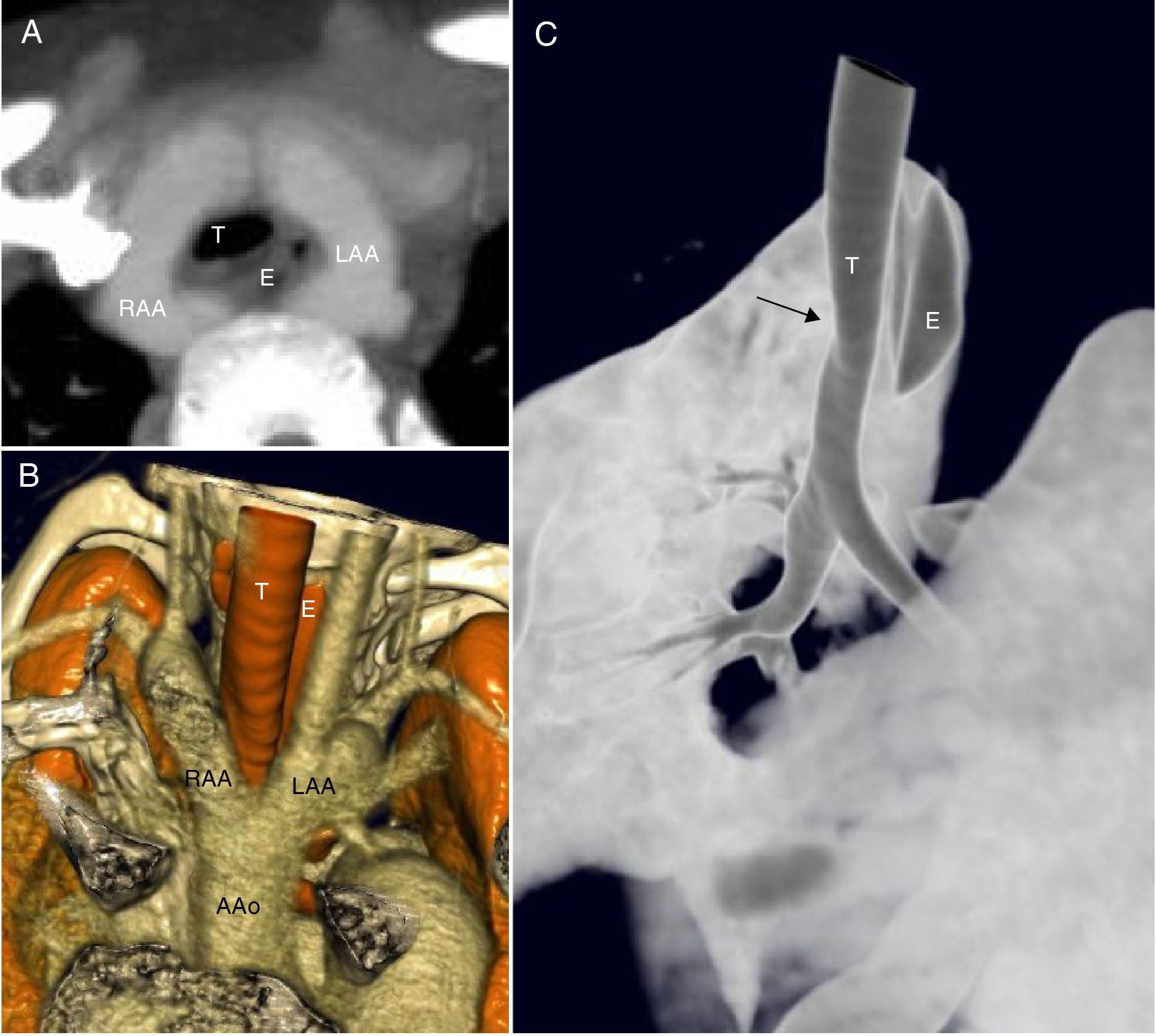
 Reconstrucción MIP. Proyección axial oblicua en donde se aprecia la relación entre el anillo vascular completo y la tráquea (T) y el esófago (E) que discurren por dentro del mismo. B) reconstrucción volumétrica de estructuras vasculares y vía aérea. C) Reconstrucción volumétrica de superficie para valorar la vía aérea. En esta imagen se aprecia la ligera impronta que produce el anillo vascular sobre la pared anterior de la tráquea, sin estenosis significativa. AAo: ascending aorta (aorta ascendente); LAA: left aortic arch (arco aórtico izquierdo); RAA: right aortic arch (arco aórtico derecho).")
A) Reconstrucción MIP. Proyección axial oblicua en donde se aprecia la relación entre el anillo vascular completo y la tráquea (T) y el esófago (E) que discurren por dentro del mismo. B) reconstrucción volumétrica de estructuras vasculares y vía aérea. C) Reconstrucción volumétrica de superficie para valorar la vía aérea. En esta imagen se aprecia la ligera impronta que produce el anillo vascular sobre la pared anterior de la tráquea, sin estenosis significativa. AAo: ascending aorta (aorta ascendente); LAA: left aortic arch (arco aórtico izquierdo); RAA: right aortic arch (arco aórtico derecho).
El RTS es causado por la deleción del cromosoma 16p13.3 (CREBBP), tiene una frecuencia de 1 en 100,000 a 125,000 recién nacidos. Se caracteriza por presentar retraso mental y en el crecimiento, obesidad, rasgos craneofaciales, cataratas, fisuras palpebrales hacia abajo, paladar alto, pulgares gruesos, alteraciones renales y defectos cardiacos congénitos1.
Las alteraciones cardiovasculares se presentan en el 17-38% de los pacientes con RTS e incluyen una gran variedad de problemas cardiacos como defectos septales, conducto arterioso, estenosis valvular pulmonar, coartación de aorta, síndrome de ventrículo izquierdo hipoplásico e hipertensión arterial pulmonar. Excepcionalmente se han descrito casos de RTS asociado con anillo vascular responsable de la obstrucción traqueoesofágica1–4.
En nuestra paciente con RTS se encuentra una asociación poco frecuente de anillo vascular completo y comunicación interventricular pequeña.
Es importante realizar el estudio anatómico prenatal y de cariotipo de deleción del cromosoma 22q11 en todos los casos de anillo vascular aislado5,6.
El anillo vascular completo se produce por la persistencia de los cuartos arcos aórticos y su unión con las aortas dorsales dando lugar a 2 arcos, uno derecho y posterior y otro izquierdo y anterior. Los anillos vasculares rodean al esófago y la tráquea dando origen a los síntomas en la deglución y respiración7.
Las pruebas que confirman el diagnóstico son las técnicas de imagen. El esofagograma muestra las identaciones esofágicas. La ecocardiografía se usa en aquellos pacientes que tengan sospecha clínica. La broncoscopia muestra la compresión extrínseca de la tráquea8,9.
Las técnicas de imagen no invasivas como la tomografía cardiaca pueden demostrar cambios en el calibre de la tráquea además de la ubicación, el grado y la extensión del estrechamiento traqueal. La resonancia magnética se ha convertido en la técnica de diagnóstico de elección; muestra la anatomía vascular, cardiaca, pulmonar y torácica, permitiendo la delimitación anatómica y la planificación de la cirugía8–10.
El manejo de los anillos vasculares asintomáticos debe ser individualizado. Los pacientes asintomáticos con anillos completos deben someterse a cirugía electiva cuando existe la progresión de la lesión de la vía aérea con el tiempo. Mientras que los anillos incompletos asintomáticos pueden mantenerse bajo vigilancia9,10.
La asociación de RTS y anillo vascular completo es poco frecuente. Es importante que ante la sospecha de anomalías cromosómicas los pacientes sean valorados por el cardiólogo pediatra para su diagnóstico. Las técnicas de imagen no invasivas son de ayuda para la delimitación de la anatomía precisa y la planificación quirúrgica.