









Las enfermedades cardiovasculares son la principal causa de morbilidad y mortalidad en los países desarrollados. Los trastornos en el metabolismo de las lipoproteínas se encuentran en el 60% de las personas con una cardiopatía isquémica (CI) y está ampliamente demostrado que el control de las concentraciones de colesterol con medidas dietéticas y/o farmacológicas disminuye la morbimortalidad cardiovascular y supone un impacto importante en la prevención cardiovascular. Sin embargo, muchas de las personas con hiperlipemias no están diagnosticadas y, por tanto, no reciben el tratamiento adecuado.
Las hiperlipemias familiares constituyen un importante problema de salud pública y se estima que al menos un 5% de la población general presenta un trastorno heredado del metabolismo de las lipoproteínas. Se caracterizan por aumento en las concentraciones plasmáticas de colesterol y/o triglicéridos, una marcada agregación familiar, un mayor riesgo de desarrollar una enfermedad cardiovascular prematura de causa arteriosclerótica y la presencia de depósitos variables de colesterol en tejidos extravasculares como la piel, los tendones y el hígado.
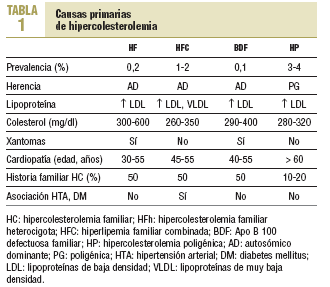
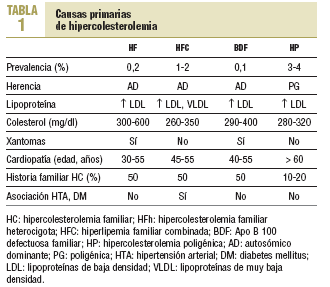
La expresión fenotípica de las hiperlipemias familiares es variable. Se distinguen: a) las que cursan con hipercolesterolemia pura (elevación del colesterol unido a lipoproteínas de baja densidad [cLDL] con el resto de los parámetros lipídicos normales), como la hipercolesterolemia familiar (HF), la Apo B-100 defectuosa familiar (BDF) y la hipercolesterolemia poligénica (HP); b) las que se presentan como hiperlipemias mixtas o combinadas, con una elevación de colesterol total y de triglicéridos que da fenotipos IIb o III, como la hiperlipemia familiar combinada (HFC) y la hiperlipoproteinemia tipo III o disbetalipoproteinemia familiar (HLP-III); c) hipertrigliceridemia pura (fenotipo I, IV o V, según la presencia de quilomicrones) (tabla 1).
En esta revisión abordaremos las hiperlipemias familiares que con frecuencia se detectan en las consultas de atención primaria.
Hipercolesterolemia familiar
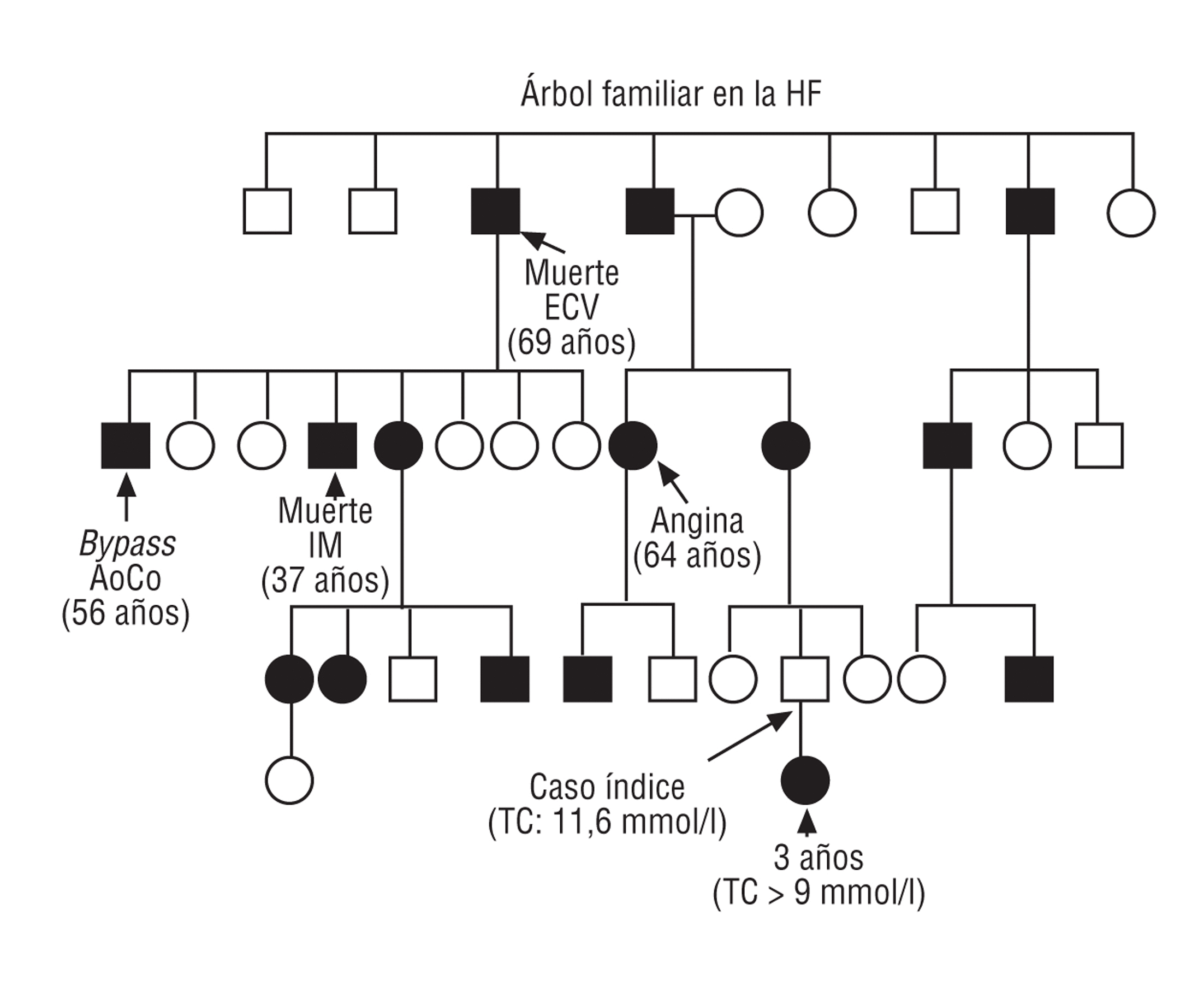
La HF está causada por mutaciones en el gen del receptor de las lipoproteínas de baja densidad (r-LDL), lo que produce una reducción importante en el número de receptores hepáticos funcionalmente activos y, por tanto, una elevación del cLDL de al menos 2 veces el valor normal. Se transmite de forma autosómica dominante con penetrancia completa1 (fig. 1). Es muy importante realizar un diagnóstico precoz de la HF debido a la elevada incidencia de enfermedad coronaria en edades tempranas de la vida, tanto en varones como en mujeres. Si los pacientes no reciben tratamiento farmacológico, aproximadamente el 75% de los varones con HFh presentará un episodio coronario antes de los 60 años de edad2,3. Además, la HF se asocia con una disminución de la esperanza de vida2,4 y la enfermedad cardiovascular es la principal causa de mortalidad5,6.
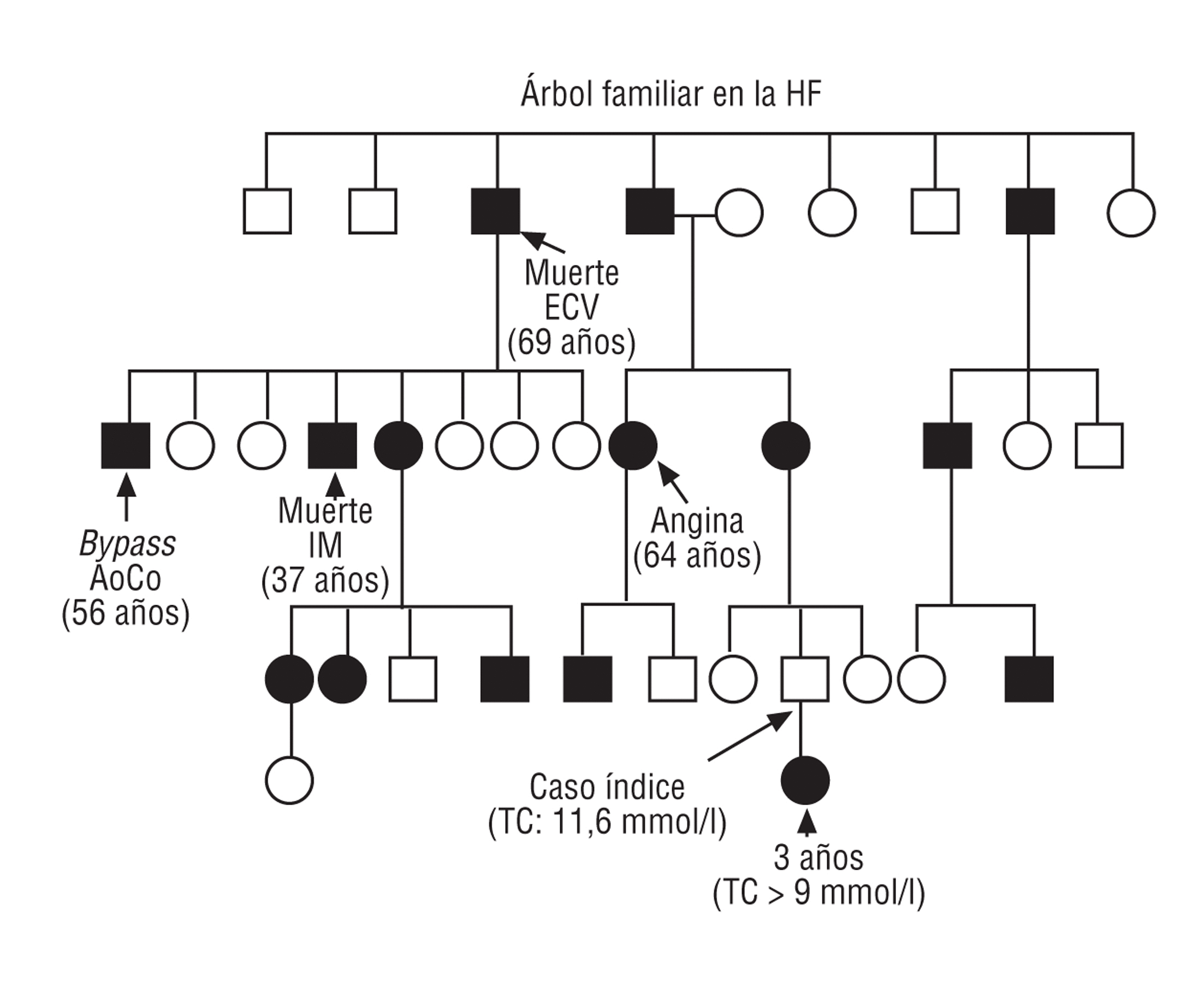
Fig. 1. Árbol familiar en la hipercolesterolemia familiar heterocigota con mutación en el gen del r-LDL (313 + 1G/A). Elaboración propia.
Se reconocen 2 variantes de la enfermedad: una heterocigota (HFh, se hereda un alelo defectuoso y el otro normal), con una incidencia de al menos 1 caso por 400-500 personas en la población general en la mayoría de los países occidentales, y otra homocigota (HFho, se heredan ambos alelos defectuosos), que es muy rara y tiene una prevalencia estimada de 1 caso por millón de habitantes.
Hipercolesterolemia familiar homocigota
Se caracteriza por presentar desde el nacimiento concentraciones extremadamente elevadas de colesterol total, que pueden alcanzar los 1.000 mg/dl. Los xantomas tendinosos, cutáneos y tuberosos, el arco corneal y la enfermedad arteriosclerótica pueden presentarse en la primera década de la vida.
Es característico el compromiso de la raíz aórtica, que produce estenosis de la válvula, acompañado de síntomas como angina, disnea y síncope con el ejercicio7,8. La muerte súbita y el infarto agudo de miocardio (IAM) antes de los 30 años de edad eran habituales hasta la aparición de nuevas medidas terapéuticas, como la LDL-aferesis.
Hipercolesterolemia familiar heterocigota
El aumento en la concentración plasmática de cLDL está presente desde el nacimiento y permanece como el único hallazgo clínico hasta la segunda década de la vida.
Hay una importante variabilidad en la concentración de cLDL de unos sujetos a otros y puede haber solapamiento con personas afectadas de otros tipos de hipercolesterolemia. Esta variabilidad se debe principalmente al tipo de mutación en el gen del receptor LDL, aunque hay otros factores que afectan a las concentraciones de LDL, como el sexo, la edad, el índice de masa corporal (IMC) y la dieta.
Dependiendo de la edad y del nivel de cLDL, son característicos de esta enfermedad los depósitos de colesterol en los tendones (xantomas), los párpados y la córnea9,10. Los sitios más frecuentes de aparición de los xantomas son los tendones aquíleos y los extensores de la mano, el codo y el rotuliano. En etapas iniciales no son visibles; por ello es importante palpar los tendones para determinar si hay irregularidades y asimetría. Ya que la palpación de los xantomas es en ocasiones dudosa, se puede utilizar la ecografía como método diagnóstico. En ocasiones puede haber dolor tendinoso que incluso puede llegar a una tendinitis recurrente. También se ha descrito una poliartritis no deformante autolimitada.
Los xantelasmas son depósitos de colesterol en los párpados que suelen ser planos y de color anaranjado. Aunque aparecen con relativa frecuencia en la HF, no son patognomónicos de ella. El valor clínico del arco corneal es mayor cuanto más joven es el sujeto que los presenta y se considera como signo de HF si aparece antes de los 45 años de edad y es completo.
Enfermedad cardiovascular en la hipercolesterolemia familiar
La importancia del diagnóstico precoz de la HF radica en la elevada incidencia de enfermedad coronaria en edades tempranas de la vida, con una antelación de unos 20 años respecto a los pacientes sin HF11-14. Estudios angiográficos en personas jóvenes con HF han demostrado la presencia de lesiones ateroscleróticas a partir de los 17 años en los varones y de los 25 en las mujeres.
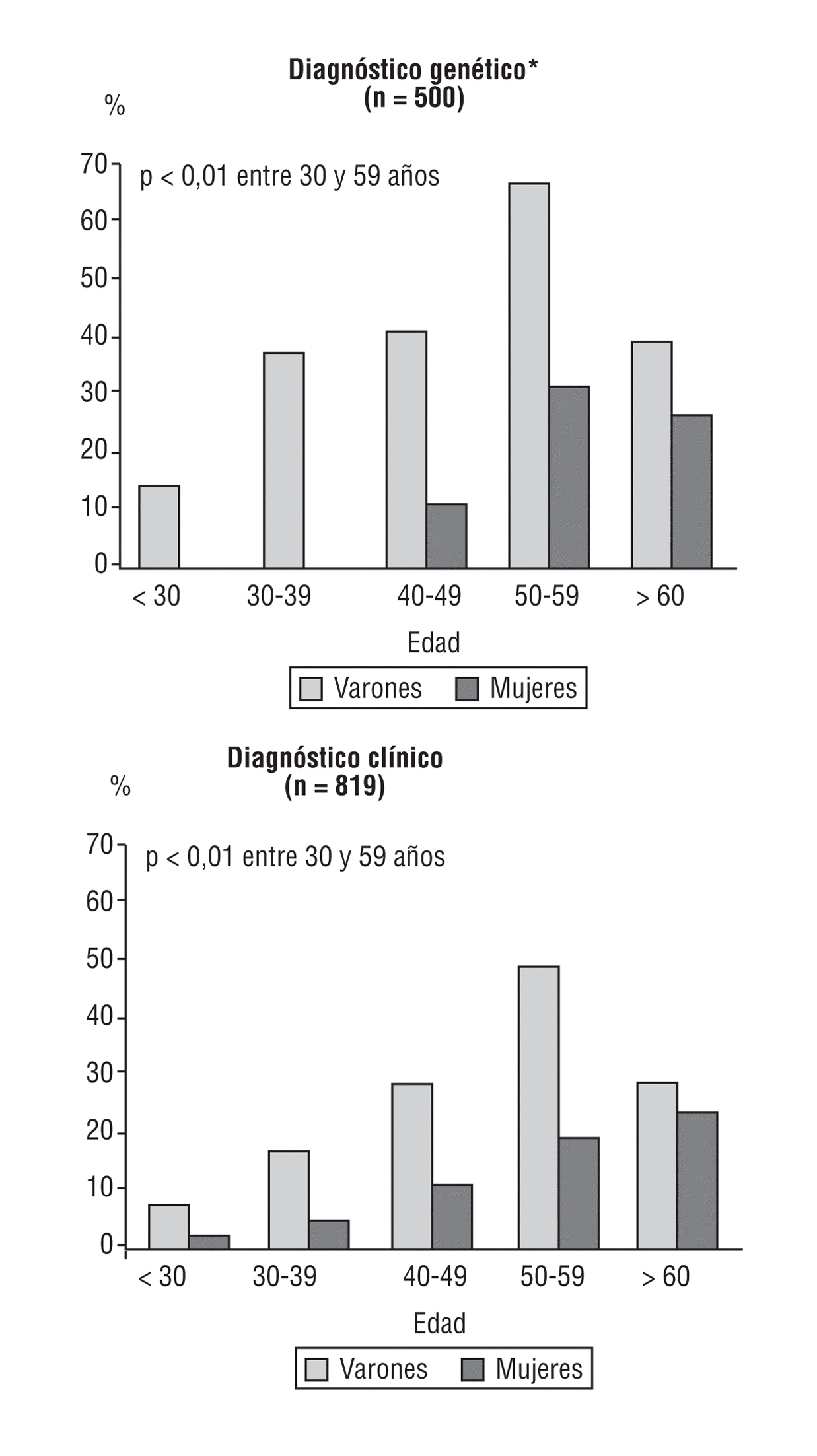
En el Registro Español de HF12, la frecuencia de cardiopatía isquémica prematura fue del 12% en las mujeres y del 27,3% en los varones. La edad media de aparición de los primeros síntomas en varones y mujeres fue a los 43 y 52 años, respectivamente. La enfermedad coronaria es más severa en los varones que en las mujeres y como primera manifestación predominan el infarto de miocardio en los varones y la angina en las mujeres. El 55% de los varones y el 24% de las mujeres con diagnóstico clínico de HF han presentado manifestaciones de enfermedad coronaria prematura en la década de los 50 años de edad (fig. 2).
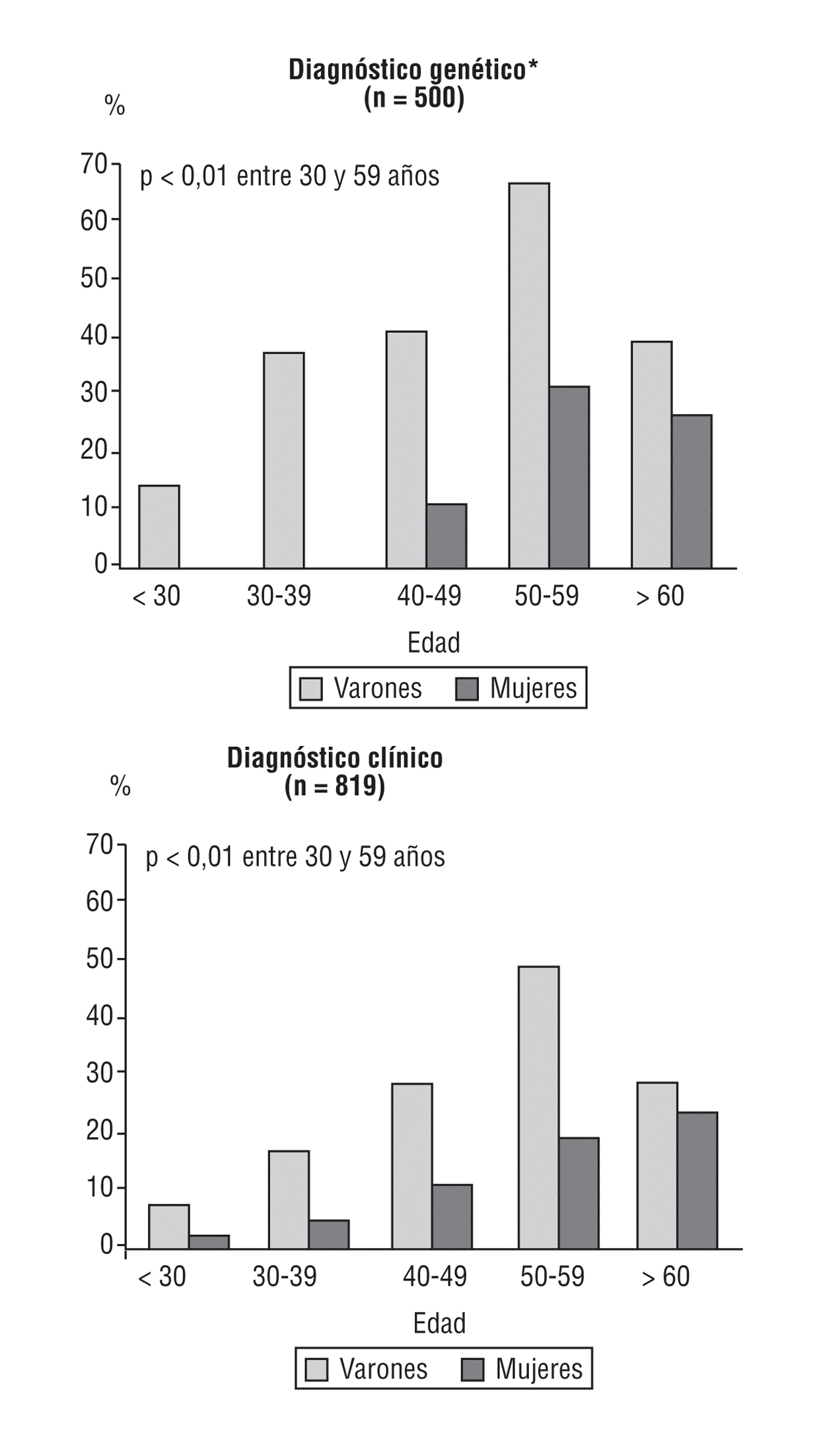
Fig. 2. Enfermedad cardiovascular en la hipercolesterolemia familiar, de acuerdo con el diagnóstico. Datos no publicados. Adaptada de Alonso et al12.
Aunque en esta población el riesgo cardiovascular (RCV) es muy elevado, hay pacientes que llegan a edades muy avanzadas sin evidencias de enfermedad cardiovascular (ECV)15. Esto sugiere que, en esta población, el desarrollo y el pronóstico de la ECV varían de manera considerable. Los factores que pueden contribuir a explicar esta variabilidad son: la mutación causal y otros polimorfismos involucrados en el metabolismo lipídico, el sexo, la edad, el tabaquismo, la dieta, la concentración de colestrol unido a lipoproteínas de alta densidad (cHDL) y los triglicéridos, entre otros12,16-18.
Diagnóstico de la hipercolesterolemia familiar
Para el diagnóstico de la HF es fundamental conocer las alteraciones lipídicas en el caso índice y especialmente en los familiares de primer grado. Una hipercolesterolemia pura en la mitad de los familiares, en especial si hay menores de 18 años afectados, sumado a los signos clínicos (xantomas tendinosos y arco corneal antes de los 45 años), apoyará el diagnóstico clínico de hipercolesterolemia familiar heterocigota19 (tabla 2). El diagnóstico definitivo se establece mediante el estudio genético del r-LDL. Sin embargo, en un 15-20% de los pacientes con diagnóstico clínico de HF (>= 8 puntos) puede no detectarse la mutación en el r-LDL porque tienen la alteración genética en otro locus.

Diagnóstico genético
En España se ha desarrollado y está disponible un método diagnóstico rápido y fiable conocido como «biochip». El chip de ADN es capaz de identificar las 185 mutaciones del r-LDL más frecuentes en la población española. Se basa en la capacidad de las moléculas de ADN para reconocer su secuencia complementaria. Básicamente, consiste en una superficie de vidrio modificada mediante procedimientos químicos en la que está depositado un gran número de secuencias génicas complementarias a cada una de las mutaciones.
Hipercolesterolemia poligénica
Es la hipercolesterolemia de causa genética más frecuente y su prevalencia se estima en un 4%. Se manifiesta a partir de la tercera década de la vida y los antecedentes familiares de hipercolesterolemia son menos frecuentes que en otras formas de hipercolesterolemia genética (10-20% de los familiares de primer grado). El colesterol suele ser de 280-320 mg/dl y no se observan xantomas.
Hiperlipemia familiar combinada
Se estima que un 1-2% de la población general está afectada. Por tanto, en España hay entre 400.000 y 600.000 personas. Aproximadamente el 20% de los sujetos con infarto de miocardio o enfermedad coronaria prematura (ECP) presentan una hiperlipemia familiar combinada (HFC), cifra que aumenta hasta un 40% cuando se consideran todos los supervivientes de un infarto de miocardio.
El mecanismo exacto de transmisión no se conoce con exactitud, pero dada la agregación familiar que se observa, parece ser efecto de un gen dominante junto a factores ambientales moduladores20,21. Generalmente, la concentración de colesterol plasmático se encuentra entre 260 y 350 mg/dl y la de triglicéridos, aunque varía mucho, entre 250 y 450 mg/dl22. A menudo se observa un valor disminuido de cHDL < 35 mg/dl. Una Apo B > 130 mg/dl es un hallazgo frecuente en la HFC y corresponde a un aumento de partículas de lipoproteínas de muy baja (VLDL) y baja (LDL) densidad en este trastorno. Las partículas de LDL suelen ser pequeñas y densas, las cuales son más aterogénicas.
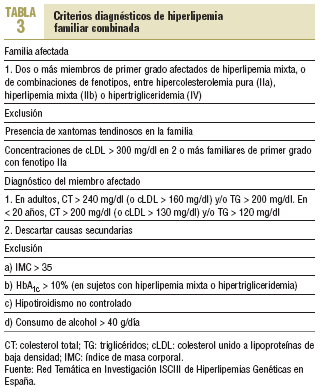
No se dispone de ninguna prueba diagnóstica de certeza para la HFC. El diagnóstico debe basarse en el estudio familiar y la exclusión de otras hiperlipemias. Hay una agregación familiar importante de la hiperlipemia, generalmente en mayores de 20 años, aunque también se puede observar en la infancia. En una misma familia hay sujetos con colesterol y/o triglicéridos elevados. Por tanto, la expresión de la hiperlipemia cambia en el propio individuo a lo largo del tiempo, y también dentro del grupo familiar22,23.
A diferencia de la HF que aparece ya en el nacimiento, la HFC se expresa totalmente al final de la segunda o al empezar la tercera década de la vida. En la HFC no hay xantomas tendinosos; sin embargo, no es infrecuente la presencia de un arco corneal prematuro. Aunque no hay una definición de consenso, la mayor parte de los estudios considera que un paciente presenta una HFC si reúne los criterios clínicos que se muestran en la tabla 3.
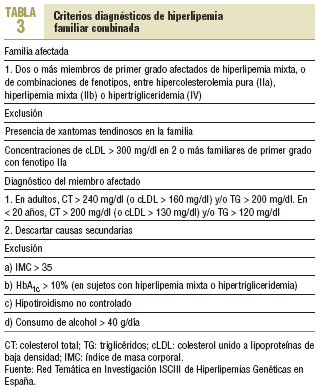
Hasta un 20-30% tiene diabetes, hipertensión arterial y obesidad de predominio central con resistencia a la insulina24. También es frecuente la infiltración grasa del hígado con una discreta elevación de las enzimas hepáticas.
Disbetalipoproteinemia familiar o hiperlipoproteinemia tipo III (HLP-III)
Se presenta como una hiperlipemia mixta grave con una elevación del colesterol y los triglicéridos plasmáticos habitualmente por encima de 350 mg/dl y con cifras similares para ambos, secundarios al aumento de lipoproteínas remanentes procedentes de los quilomicrones y las VLDL. Se requiere la presencia de un genotipo E2/E2 y de algún otro factor precipitante como diabetes, obesidad, hipotiroidismo, etc. Se transmite de forma autosómica recesiva incompleta y con baja penetrancia25,26. Se estima que 1 de cada 5.000 personas en la población general expresa la enfermedad, y aproximadamente el 1% de los sujetos con enfermedad coronaria prematura presenta esta hiperlipemia. Es rara en niños y en las mujeres antes de la menopausia. En la HLP-III es patognomónica la presencia de depósitos lipídicos en los surcos de las palmas de las manos conocidos como xantomas palmares.
La cardiopatía isquémica es la causa principal de muerte en la HLP-III, y en el momento del diagnóstico, el 50% de los sujetos tiene síntomas en los territorios coronarios, cerebrovasculares y en las extremidades inferiores27.
Tratamiento de las hiperlipemias hereditarias
Las tablas de cálculo de riesgo cardiovascular no se deben utilizar en estos pacientes, ya que se consideran de muy alto riesgo cardiovascular28. La mayoría de ellos requerirá tratamiento farmacológico y éste se puede iniciar a la vez que las medidas higiénico-dietéticas, ya que el efecto de estas últimas sobre el cLDL será discreto. Además del tratamiento hipolipemiante hay que valorar la presencia de otros factores de riesgo cardiovascular.
La elección del fármaco hipolipemiante dependerá del trastorno lipídico predominante, del riesgo cardiovascular global, de su espectro de acción, de sus características farmacológicas y de sus potenciales efectos adversos. El tratamiento hipolipemiante es crónico y sólo debe suspenderse ante la presencia de efectos adversos o intolerancia al fármaco.
Inhibidores de la hidroximetil glutaril CoA reductasa
(estatinas)
Las estatinas son eficaces en la reducción del colesterol total y el cLDL y tienen una buena tolerancia. Además, se ha demostrado que disminuyen la morbimortalidad de causa coronaria y el ictus en pacientes en prevención primaria de alto riesgo y en pacientes en prevención secundaria29.
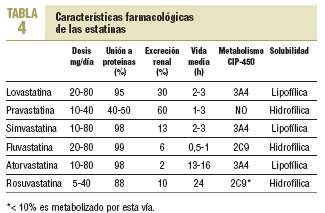
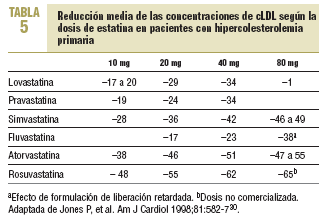
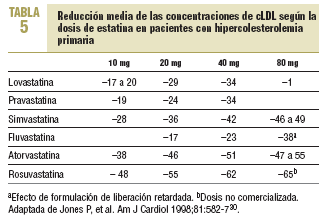
Las estatinas difieren en una serie de parámetros farmacocinéticos (tabla 4). Su efecto terapéutico hipocolesterolemiante es similar en todos cuando se utilizan dosis equivalentes. Según el tipo y la dosis empleada, se consiguen reducciones del cLDL de hasta un 60%. El orden de potencia de las estatinas cuando se comparan por miligramos es: rosuvastatina > atorvastatina > simvastatina > pravastatina = lovastatina > fluvastatina. Los cambios en el cLDL son independientes de sus valores basales. Al doblar la dosis de cualquier estatina, el aumento relativo en la reducción de cLDL es en general del 6-8% (tabla 5)30. Además, no se ha evidenciado taquifilaxia o tolerancia con el tratamiento crónico con estatinas. El efecto sobre los triglicéridos es dependiente de su concentración basal y es mayor en los sujetos con triglicéridos > 250 mg/dl. El efecto sobre el cHDL parece depender también de los triglicéridos basales y de la dosis, a excepción de atorvastatina, que con dosis altas puede producir una disminución del cHDL.
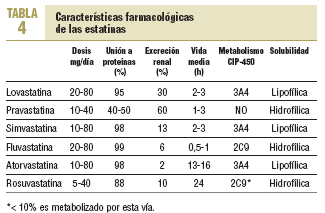
Aparte de sus efectos sobre el perfil lipídico, se han descrito otras acciones beneficiosas de las estatinas sobre la pared arterial. Estos efectos se conocen como efectos pleiotrópicos y explicarían el beneficio adicional no atribuible a la reducción del cLDL observado en muchos estudios de intervención.
Interacción con otros medicamentos
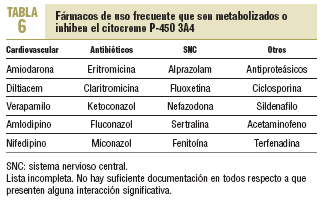
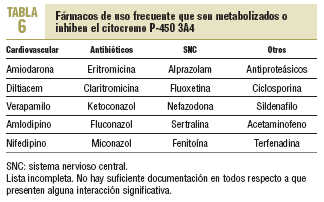
La mayoría de las interacciones no tiene una relevancia clínica importante. Sin embargo, en un pequeño porcentaje de los sujetos que toman estatinas se producen alteraciones hepáticas y musculares que pueden ser graves en algunos de ellos. La mayoría de los pacientes están polimedicados o deben usar algún fármaco de manera ocasional, lo que aumenta la posibilidad de interacción. El citocromo P-450 3A4 es el causante del metabolismo de más del 50% de los fármacos utilizados (tabla 6). Por el momento, no es posible predecir las interacciones farmacológicas con relevancia clínica.
Los efectos adversos más frecuentes son el estreñimiento, la dispepsia, las náuseas, la cefalea y el dolor gastrointestinal, que suelen ser transitorios y leves. En < 1% de los casos se produce un aumento de las transaminasas (más de 3 veces el valor normal) que es dependiente de la dosis y similar para todas las estatinas. El efecto adverso más grave está relacionado con la afección muscular, que puede manifestarse como mialgia, miositis, miopatía y rabdomiólisis. En general, la afección más frecuente es la mialgia sin elevación de la creatinfosfocinasa (CPK). La miopatía y la rabdomiólisis son muy raras (< 1% para la primera y < 0,1% para la segunda). Hay una serie de factores que predisponen a un mayor riesgo de miopatía con el tratamiento con estatinas (tabla 7).

Se recomienda realizar un primer control a partir de las 8 semanas de instaurar el fármaco para valorar las transaminasas y la creatinfosfocinasa si hay clínica muscular. Si es necesario aumentar la dosis o asociar un segundo fármaco hipolipemiante, se debe realizar un nuevo control a las 8 semanas del cambio. Ante la presencia de síntomas musculares hay que valorar los factores desencadenantes y hacer las determinaciones de CPK. En algunos casos, aun ante valores de CPK normales, es necesario suspender la medicación por el dolor muscular.
Resinas secuestradoras de ácidos biliares
Las resinas actúan a nivel local como secuestradoras de ácidos biliares en el intestino.
El colesterol total puede disminuir entre un 10 y un 25% a expensas del cLDL que se reduce un 15-30% según la dosis utilizada. El 75% del efecto hipolipemiante se consigue con dosis medias (aproximadamente, 12 g de colestiramina o 15 g de colestipol). El cHDL puede aumentar discretamente un 3-5% y los triglicéridos un 10%. En la HF son el fármaco de primera elección en niños y adolescentes. En adultos suele utilizarse en tratamiento combinado con estatinas.
Los principales efectos adversos de las resinas son gastrointestinales. Se recomienda que cualquier medicamento que deba administrarse a pacientes que reciban resinas se tome 1 h antes o bien 4 h después.
Inhibidores de la absorción del colesterol
La ezetimiba es la primera de una nueva clase de moléculas que inhiben selectivamente la absorción del colesterol de la dieta y de origen biliar en el intestino31. Actúa en las microvellosidades intestinales, donde inhibe una molécula que se cree es un transportador de colesterol. No afecta a la absorción de triglicéridos y de las vitaminas liposolubles. Tiene una vida media larga, de 22 h, lo que permite su administración en una sola dosis diaria. No es metabolizada por el citocromo P-450. En terapia combinada, la adición de ezetimiba al tratamiento con estatinas produce una reducción adicional del 21% sobre el efecto de la estatina sola, con independencia de la estatina utilizada. Por ello, la mayoría de los pacientes logra alcanzar el objetivo de cLDL32. Además, se ha demostrado que el efecto reductor del cLDL de la ezetimiba, junto con la dosis más baja de la estatina (10 mg), es similar a la obtenida con la dosis más alta de la estatina sola (hasta 80 mg/día) y con una buena tolerancia.
Derivados del ácido fíbrico (fibratos)
Son el tratamiento de elección cuando se trata de una hipertrigliceridemia, excepto en la hiperquilomicronemia tipo I, en la cual los fibratos no están indicados, ya que el tratamiento es exclusivamente dietético. Los pacientes con valores de triglicéridos elevados y de cHDL bajos obtienen el mayor beneficio con el tratamiento con fibratos.
Los fibratos son agonistas de unos receptores nucleares, los PPAR-*33(peroxisome proliferator-activated receptor), cuya estimulación reduce las concentraciones de VLDL mediante la disminución de su producción y el aumento de su eliminación34. El efecto de los fibratos de última generación sobre el metabolismo del colesterol es moderado ya que actúan discretamente sobre la HMG-CoA reductasa, promoviendo la eliminación biliar de colesterol.
La reducción de los triglicéridos puede superar el 50% en las hipertrigliceridemias y la de cLDL alcanza el 25% con los fibratos de última generación (fenofibrato, ciprofibrato). Pueden aumentar las concentraciones de cHDL hasta un 20%, especialmente en los casos con un cHDL < 40 mg/dl32. En general, los fibratos son seguros y su tolerancia es buena.
Los efectos secundarios más frecuentes son las molestias digestivas, seguidas de la cefalea, los trastornos del sueño, las mialgias y las erupciones cutáneas. La hepatitis y la miositis son excepcionales.
Los fibratos pueden potenciar el efecto de los anticoagulantes orales, por lo que se debe controlar el tiempo de protrombina cuando se usan ambos fármacos.
Tratamiento combinado
En ocasiones es necesario utilizar el tratamiento combinado con diferentes fármacos en las hiperlipemias familiares graves que no alcanzan los objetivos del tratamiento con monoterapia o en los pacientes que no toleran altas dosis de un único fármaco35.
Las combinaciones de fármacos tienen diferentes efectos en el perfil lipídico:
1. Estatinas y fibratos: reducción del cLDL hasta el 46% con fenofibrato y del 35-57% de los triglicéridos. El aumento del cHDL fluctúa entre el 12 y el 22%, según la combinación utilizada. Está contraindicada la asociación de gemfibrocilo con estatinas.
2. Estatinas y resinas: reducción del cLDL de hasta el 50%. Pueden aumentar los triglicéridos, por lo que no se recomienda esta asociación en pacientes con hiperlipemias mixtas.
3. Estatinas y ezetimiba: reducción del cLDL hasta un 65% y de los triglicéridos del 8-10%.
Tratamiento según tipo de hiperlipemia
Hipercolesterolemia familiar. Prácticamente todos los pacientes necesitan tratamiento farmacológico con estatinas. En la mayoría de los adultos con HF es preciso reducir el cLDL > 45% para alcanzar el objetivo terapéutico. Si con dosis medias o altas no se consigue el objetivo en el cLDL, se pueden añadir resinas o ezetimiba.
Hiperlipemia familiar combinada o mixta. Si los triglicéridos son < 300 mg/dl, las estatinas son el primer fármaco de elección. Sin embargo, si los triglicéridos no disminuyen hasta un nivel óptimo y los valores de cHDL permanecen bajos, aun cuando se consiga el objetivo en el cLDL, se puede añadir un fibrato.
En todo tratamiento combinado, el incremento de las dosis de estatinas se debe hacer de forma paulatina y con controles periódicos de las transaminasas y la CPK. La asociación estatina-fibrato puede ser bien tolerada si se excluye a los pacientes con insuficiencia renal o que estén tomando otra medicación de forma crónica, como la terapia inmunodepresora. Si se utilizan dosis bajas de cada uno de los fármacos y se separan las tomas de cada uno 12 h, las reacciones adversas severas con la combinación de estatinas y fibratos no son un problema mayor en la práctica clínica habitual.
Hipercolesterolemia familiar en niños. En los niños con HF, el inicio del tratamiento farmacológico dependerá del riesgo cardiovascular (historia familiar de enfermedad coronaria prematura), del sexo del niño y de las concentraciones de colesterol total después de realizar una dieta adecuada. En general, el tratamiento suele comenzarse a partir de los 10 años y, por el momento, el fármaco de elección es una resina. Recientemente, la Food and Drug Administration ha aprobado el uso de estatinas en varones con HF a partir de los 10 años, y en las niñas a partir de 1 año después de la menarquia. Las estatinas han sido utilizadas en niños en estudios a largo plazo y no se ha demostrado que interfieran con el desarrollo ponderal, estatural ni gonadal. Hay que recordar que las mujeres en edad fértil deben suspender las estatinas si desean tener un embarazo. En la mayoría de los casos, no se recomienda el uso de estatinas antes de los 18 años de edad36,37.
Hipercolesterolemia familiar grave. En los casos con HF muy grave, especialmente en los que presentan una HF homocigota o una HF heterocigota resistente al tratamiento farmacológico (cLDL > 300 mg/dl sin evidencia clínica de enfermedad aterosclerótica, o se trata de una HF con cardiopatía isquémica y con un cLDL > 200 mg/dl después del tratamiento farmacológico) será preciso utilizar la LDL-aféresis. Durante el uso de la LDL-aféresis se debe continuar el tratamiento farmacológico.
